Baricitinib (Olumiant®) ▼
Neue Indikation: atopische Dermatitis
Zugelassene Indikation und Wirkmechanismus
Olumiant® (Baricitinib) wurde 2017 zur Behandlung von mittelschwerer bis schwerer aktiver rheumatoider Arthritis bei erwachsenen Patienten zugelassen, die auf eine vorangegangene Behandlung mit einem oder mehreren krankheitsmodifizierenden Antirheumatika (DMARD) unzureichend angesprochen oder diese nicht vertragen haben. Im November 2020 erfolgte die Indikationserweiterung zur Behandlung von mittelschwerer bis schwerer atopischer Dermatitis bei erwachsenen Patienten, die für eine systemische Therapie infrage kommen.
Baricitinib ist ein selektiver und reversibler Inhibitor von Januskinase (JAK)1 und JAK2. JAK sind Enzyme, die intrazelluläre Signale von Zelloberflächenrezeptoren für eine Reihe von Zytokinen und Wachstumsfaktoren weiterleiten, die an Hämatopoese, Entzündung und Immunabwehr beteiligt sind. Innerhalb des intrazellulären Signalweges phosphorylieren und aktivieren JAK Signaltransduktoren und Aktivatoren der Transkription (STAT), die wiederum die Genexpression innerhalb der Zelle aktivieren. Baricitinib moduliert diese Signalwege, indem es die enzymatische Aktivität von JAK1 und JAK2 teilweise hemmt und damit die Phosphorylierung und Aktivierung von STAT reduziert.
Markteinführung
Olumiant® (Baricitinib) ist seit 2017 auf dem deutschen Markt.
Bewertung
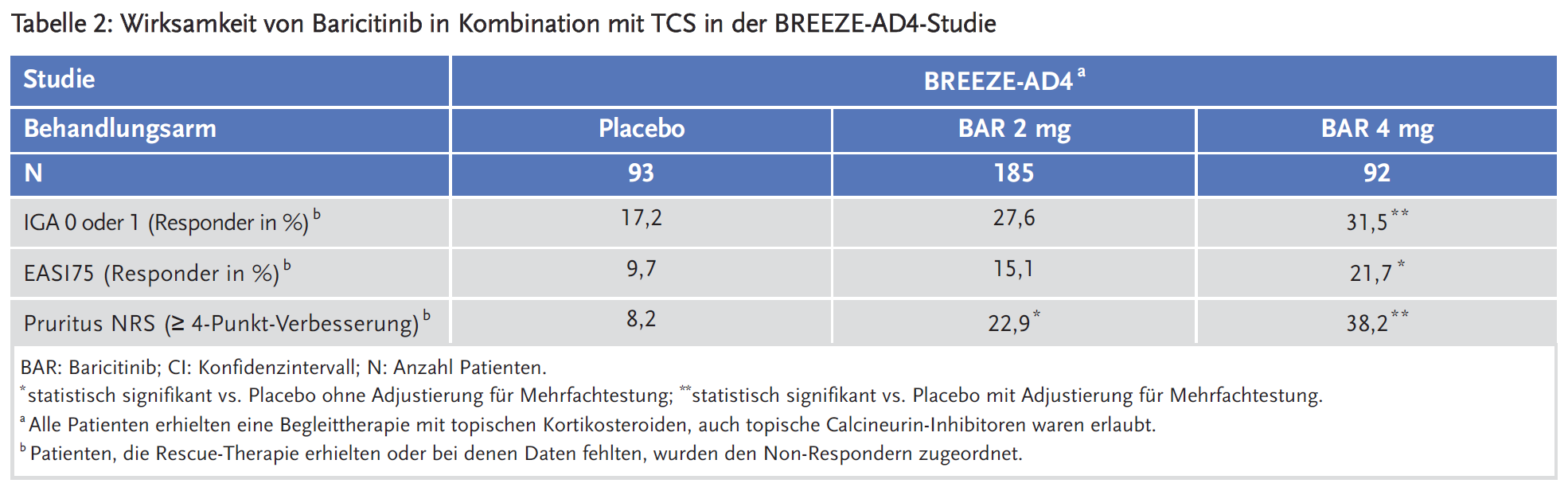
Baricitinib zeigte in den Zulassungsstudien über 16 Wochen einen statistisch signifikanten Vorteil im Vergleich zu Placebo in der Behandlung von mittelschwerer bis schwerer atopischer Dermatitis bei erwachsenen Patienten, die für eine systemische Therapie infrage kommen. Der Effekt war konsistent in der Monotherapie und in der Kombination mit topischen Kortikosteroiden (TCS). Auch bei Patienten, die ein Versagen, eine Unverträglichkeit oder eine Kontraindikation gegen eine orale Ciclosporin-Behandlung aufwiesen, war Baricitinib in Kombination mit TSC wirksamer als Placebo.
Zu den häufigsten Nebenwirkungen von Baricitinib gehören ein Anstieg des LDL-Cholesterins, Infektionen der oberen Atemwege und Kopfschmerzen. Das Sicherheitsprofil bei atopischer Dermatitis entspricht dem in der Behandlung von rheumatoider Arthritis.
Die Wirksamkeit und Sicherheit gegenüber anderen systemischen Therapien wie systemische Kortikosteroide, Ciclosporin, Azathioprin, Mycophenolatmofetil, Methotrexat oder Dupilumab kann derzeit nicht beurteilt werden, weil dazu keine Studiendaten vorliegen.
Wirksamkeit in den Zulassungsstudien
Für die Zulassung wurden drei 16-wöchige, multizentrische, doppelblinde, randomisierte, placebokontrollierte Studien (BREEZE-AD1, BREEZE-AD2 und BREEZE-AD7) durchgeführt, in denen Baricitinib in der Dosierung 1 mg, 2 mg und 4 mg einmal täglich evaluiert wurde. Die Studien schlossen 1568 erwachsene Patienten mit mittelschwerer bis schwerer atopischer Dermatitis ein, die definiert wurde als ein Wert ≥ 3 gemäß Investigator’s Global Assessment (IGA), ein Wert ≥ 16 gemäß Eczema Area and Severity Index (EASI) und eine betroffene Körperoberfläche (Body Surface Area, BSA) ≥ 10 %. Die Patienten wiesen ein unzureichendes Ansprechen oder eine Unverträglichkeit gegenüber topischer Therapie auf.
Der primäre Endpunkt war der Anteil der Patienten, die zu Woche 16 ein IGA-Ansprechen von 0 oder 1 mit einer Verbesserung von ≥ 2 Punkten erreicht haben. Als sekundäre Endpunkte wurden u. a. erhoben das EASI75- und EASI90-Ansprechen und die Verbesserung des wöchentlichen Durchschnittswerts gemäß Pruritus Numerical Rating Scale (NRS).
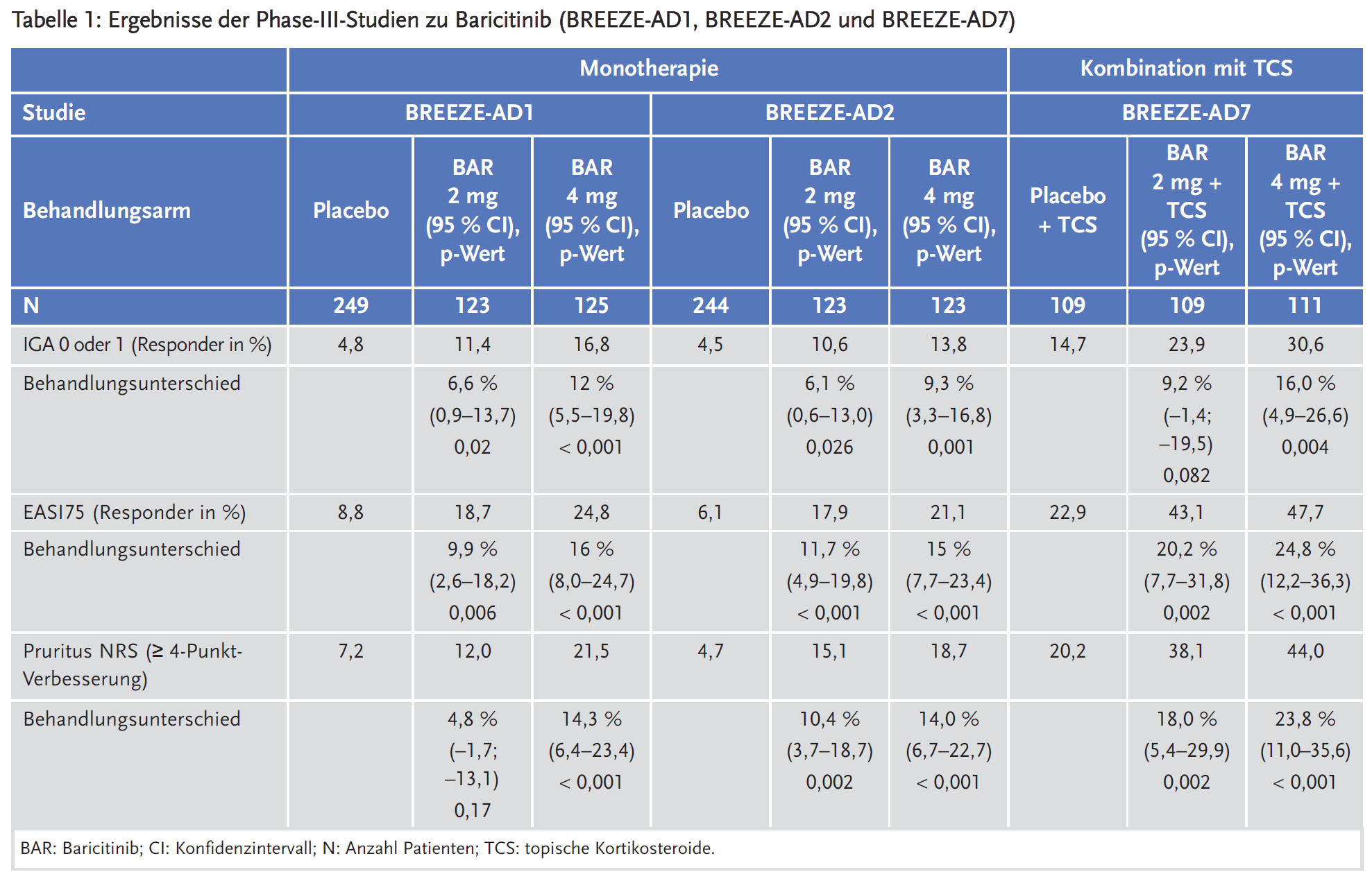
Zudem wurde eine doppelblinde, randomisierte, placebokontrollierte Langzeitstudie BREEZE-AD4 über 52 Wochen durchgeführt, um bei 463 Patienten mit einer mittelschweren bis schweren atopischen Dermatitis, die ein Versagen (n = 173), eine Unverträglichkeit (n = 75) oder eine Kontraindikation (n = 126) gegen eine orale Ciclosporin-Behandlung aufwiesen, Baricitinib in Kombination mit topischen Kortikosteroiden (TCS) zu untersuchen. Der primäre Endpunkt war der Anteil an Patienten, die ein EASI-75-Ansprechen in Woche 16 erreichten.
Ausgewählte Nebenwirkungen
Sehr häufige Nebenwirkungen unter Baricitinib sind Infektionen der oberen Atemwege und Hypercholesterinämie. Häufig treten Herpes zoster, Herpes simplex, Gastroenteritis, Harnwegsinfektionen, Pneumonie, Thrombozytose, Kopfschmerzen, Übelkeit und Bauchschmerzen auf. Gelegentliche Nebenwirkungen sind Neutropenie, tiefe Venenthrombose, Lungenembolie und Hypertriglyzeridämie.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Baricitinib ist einmal täglich unabhängig von den Mahlzeiten und der Tageszeit einzunehmen.
- Die empfohlene Dosis beträgt 4 mg einmal täglich. Eine Dosis von 2 mg täglich ist für Patienten ab 75 Jahren bzw. für Patienten mit chronischen bzw. wiederkehrenden Infekten in der Vorgeschichte angebracht. Auch für Patienten, die mit 4 mg täglich eine anhaltende Kontrolle über die Krankheitsaktivität erreicht haben und die für eine Dosisreduktion infrage kommen, kann eine Dosierung von 2 mg täglich in Betracht gezogen werden.
- Bei Patienten mit einer absoluten Lymphozytenzahl (ALC) von weniger als 0,5 x 109 Zellen/l, einer absoluten Neutrophilenzahl (ANC) von weniger als 1 x 109 Zellen/l oder einem Hämoglobinwert unter 8 g/dl sollte eine Therapie nicht begonnen werden.
- Bei Patienten mit einer Kreatinin-Clearance zwischen 30 und 60 ml/min beträgt die empfohlene Dosis 2 mg einmal täglich. Bei Patienten mit einer Kreatinin-Clearance < 30 ml/min wird die Anwendung nicht empfohlen.
- Bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Bei Patienten mit schwerer Leberfunktionsstörung wird die Anwendung nicht empfohlen.
- Bei Patienten, die gleichzeitig Inhibitoren von Organischen Anionen-Transportern vom Typ 3 (OAT3) wie etwa Probenecid, Leflunomid und Teriflunomid anwenden, beträgt die empfohlene Dosis 2 mg einmal täglich.
- Die klinische Erfahrung bei Patienten ≥ 75 Jahren ist sehr begrenzt. Für diese Patienten ist die empfohlene Dosierung 2 mg täglich.
Schulungsmaterial
Für einzelne Arzneimittel wird bereits bei der Zulassung angeordnet, dass das Arzneimittel nur unter Verwendung von Schulungsmaterialien in Verkehr gebracht werden darf. Das Schulungsmaterial dient dazu, die Wissensvermittlung zu optimieren und Hilfe bei der sicheren Anwendung des Arzneimittels zu geben, gegebenenfalls unter Einbeziehung einer patientenbezogenen Ansprache. Das behördlich beauflagte und genehmigte Schulungsmaterial zu Olumiant® besteht aus einer Broschüre für den Arzt und einem Therapiepass für Patienten.

Weiterführende Informationen
Das IQWiG wurde am 15.11.2020 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Olumiant®, erschienen am 27. Oktober 2020. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 26. November 2020 vorab online veröffentlicht.