Tixagevimab/Cilgavimab (Evusheld®) ▼
Neue Arzneimittel
Dieser Artikel wurde am 13. Juli 2022 vorab online veröffentlicht.
Zugelassene Indikation und Wirkmechanismus
Evusheld® (Tixagevimab/Cilgavimab) ist zugelassen zur Präexpositionsprophylaxe einer Coronavirus-19-Erkrankung („coronavirus disease 2019“, COVID-19) bei Erwachsenen und Jugendlichen ab 12 Jahren mit mindestens 40 kg Körpergewicht.
Tixagevimab und Cilgavimab sind monoklonale IgG1-Antikörper, die an zwei verschiedene Stellen des Spike-Proteins von SARS-CoV-2 binden. Hierdurch wird die Interaktion von SARS-CoV-2 mit dem ACE2-Rezeptor gestört und somit der Viruseintritt in die Wirtszellen behindert. Tixagevimab und Cilgavimab haben aufgrund von Aminosäuresubstitutionen in den Fc-Regionen eine verlängerte Halbwertszeit. Dies soll eine Wirksamkeit von mindestens sechs Monaten ermöglichen.
Markteinführung
Tixagevimab/Cilgavimab wurde am 25. März 2022 durch die Europäische Arzneimittel-Agentur (EMA) zugelassen. Zunächst wurde diese Kombination aus monoklonalen Antikörpern (Evusheld®) vom Bundesgesundheitsministerium (BMG) beschafft und über ausgewählte Apotheken, sogenannte Satellitenapotheken, verteilt. Am 31. Mai 2022 wurde die Präexpositionsprophylaxe mit monoklonalen Antikörpern zum Schutz vor COVID-19 in den Leistungskatalog der GKV (Gesetzlichen Krankenversicherung) für bestimmte Patientengruppen aufgenommen (1).
Bewertung
Die Zulassungsstudie PROVENT untersuchte, ob die präventive Gabe von Tixagevimab/Cilgavimab die Häufigkeit symptomatischer SARS-CoV-2-Infektionen beeinflusst (2). Eingeschlossen wurden Patienten, die zu Studienbeginn weder gegen COVID-19 geimpft noch von COVID-19 genesen waren. Die primäre Auswertung erfolgte nach 83 Tagen. Tixagevimab/Cilgavimab reduzierte signifikant die Häufigkeit symptomatischer, mittels PCR-Test bestätigter SARS-CoV-2-Infektionen (relative Risikoreduktion 77 %, Patienten mit Ereignis 0,2 % vs. 1,0 %). Eine explorative Auswertung nach sechs Monaten ergab eine vergleichbare relative Risikoreduktion. Als primärer Endpunkt wurden überwiegend milde Krankheitsverläufe registriert. Aufgrund der sehr kleinen Fallzahl ist derzeit unklar, ob die präventive Gabe von Tixagevimab/Cilgavimab auch schwere COVID-19-Verläufe verhindert, die eine stationäre Behandlung erforderlich machen oder zum Tod führen.
Die EMA schränkte die Zulassung zur Präexpositionsprophylaxe nicht auf bestimmte Indikationen ein. Die Kosten für Tixagevimab/Cilgavimab müssen gemäß Verordnung des BMG allerdings nur dann von der GKV übernommen werden, wenn entweder durch eine Impfung keine ausreichende Immunität gegen SARS-CoV-2 erzielt werden kann oder wenn Impfungen gegen COVID-19 kontraindiziert sind und zugleich ein Risiko für einen schweren Verlauf besteht (1). Mit ähnlichen Einschränkungen wurde Tixagevimab/Cilgavimab in den USA durch die FDA (U. S. Food and Drug Administration) zugelassen (3). Auch aus Sicht der AkdÄ sollte die präventive Gabe von Tixagevimab/Cilgavimab auf die genannten Patientengruppen beschränkt sein.
Aktuell fehlen klare Kriterien, wann keine „ausreichende“ Immunität nach COVID-19-Impfung besteht. Das Robert Koch-Institut (RKI) empfiehlt nur bei wenigen Erkrankungen bzw. immunsuppressiven Therapien eine serologische Überprüfung der Impfantwort. Der Antikörpertiter wird bei diesen Patienten unmittelbar vor und vier Wochen nach der dritten Impfung bestimmt (4). Die Bewertung der serologischen Diagnostik ist schwierig, da noch nicht abschließend geklärt ist, ab welcher Antikörperkonzentration von einem sicheren Schutz ausgegangen werden kann. In einer Auswertung der Zulassungsstudie von Vaxzevria (5) korrelierte ein Anti-Spike-IgG Titer von > 29 BAU/ml mit einem 50-prozentigen Schutz vor symptomatischer Infektion (6). Neben der humoralen Immunität (neutralisierende Antikörper) spielen jedoch auch das angeborene und das spezifische zelluläre Immunsystem eine wichtige Rolle bei dem Schutz vor Infektion und Erkrankung (4).
Die Studienergebnisse von PROVENT lassen sich nur eingeschränkt auf die Versorgung übertragen. Patienten mit Immundefizienz waren in der Studie PROVENT stark unterrepräsentiert (< 4 %) und Patienten mit stattgehabter COVID-19-Impfung wurden ausgeschlossen. Die Hauptzielgruppe – immunsupprimierte Patienten mit unzureichendem Ansprechen auf die Impfung – ist somit in der Studie PROVENT nicht abgebildet. Darüber hinaus ist die zweite Zielgruppe – Personen mit Kontraindikationen gegen eine COVID-19-Impfung – zumeist nicht immunnaiv, wenn für sie eine präventive Gabe von Tixagevimab/Cilgavimab infrage kommt, da frühere schwere Unverträglichkeitsreaktionen (z. B. Anaphylaxie) die häufigste Kontraindikation für eine COVID-19-Impfung sind. Patienten mit Kontraindikationen haben deshalb in der Regel mindestens eine Impfung gegen COVID-19 erhalten, bevor eine präventive Gabe von Tixagevimab/Cilgavimab erwogen wird.
Als die Studie PROVENT durchgeführt wurde, hatten sich noch nicht die Omikron-Varianten ausgebreitet. Klinische Daten aus randomisierten Studien für die Wirksamkeit von Tixagevimab/Cilgavimab gegenüber Omikron-Varianten liegen deshalb nicht vor. Labortests zeigen eine gering bis moderat reduzierte Neutralisierungsaktivität gegenüber Omikron (7). Basierend auf diesen Ergebnissen empfiehlt die FDA eine doppelt so hohe Dosis von Tixagevimab/Cilgavimab wie in der Zulassungsstudie PROVENT (jeweils 300 mg) (3). Die EMA rät dagegen aufgrund fehlender Sicherheitsdaten von einer Dosiserhöhung ab.
Insgesamt bestehen deshalb aus Sicht der AkdÄ derzeit erhebliche Unsicherheiten, ob und in welchem Maße Tixagevimab/Cilgavimab in den Zielgruppen patientenrelevante, d. h. schwere Infektionen durch die aktuell dominierenden Varianten von SARS-CoV-2 verhindert. Zudem kann ein erhöhtes kardiovaskuläres Risiko durch Tixagevimab/Cilgavimab nicht ausgeschlossen werden. Die präventive Gabe von Tixagevimab/Cilgavimab ist nach Einschätzung der AkdÄ allenfalls eine Option für Hochrisikopatienten, bei denen ein unzureichender Immunschutz durch die erfolgten COVID-19-Impfungen anzunehmen ist.
Wirksamkeit in den Zulassungsstudien
Die Zulassung von Tixagevimab/Cilgavimab basiert auf der doppelblinden, randomisierten, placebokontrollierten Phase-III-Studie PROVENT. Die Studienteilnehmer wurden im Verhältnis 1:2 zu Placebo (n = 1737) oder Tixagevimab/Cilgavimab (n = 3460) randomisiert. Die einmalige Anwendung erfolgte als zwei separate intramuskuläre Injektionen (zweimalige Injektion von Placebo bzw. jeweils einmalige Injektion von Tixagevimab und Cilgavimab). Die Studie PROVENT ist aktuell nicht abgeschlossen. Weitere klinische und pharmakokinetische Daten sollen bis zwölf Monate nach Randomisierung erhoben werden.
Die Studie PROVENT wurde in Europa (Belgien, Frankreich, Spanien, Großbritannien) und in den USA durchgeführt. Männer und Frauen wurden annährend zu gleichen Teilen eingeschlossen. Die Patienten waren im Median 54 Jahre alt. Bei 78 % lagen individuelle Risikofaktoren für einen schweren Verlauf von COVID-19 vor, vor allem Adipositas (42 %), gefolgt von Hypertonus und Rauchen. Nur wenige Patienten mit immunsuppressiver Therapie oder Immundefekten nahmen an der Studie teil (3,3 % bzw. 0,5 %).
Es wurden überwiegend (73 %) Patienten mit einem erhöhten Risiko für ein inadäquates Ansprechen auf COVID-19-Impfungen eingeschlossen. Als Risikofaktoren galten unter anderem ein Alter ≥ 60 Jahre, Adipositas, Immunsuppression und internistische Vorerkrankungen (Herzinsuffizienz, COPD, chronische Nieren- oder Lebererkrankungen). Eine Studienteilnahme war unabhängig von individuellen Risikofaktoren auch dann möglich, wenn ein erhöhtes Risiko für eine Infektion mit SARS-CoV-2 bestand, beispielsweise aufgrund einer Arbeit im medizinischen Bereich oder einer Unterbringung in Gemeinschaftsunterkünften. Es wurden nur Personen untersucht, bei denen keine COVID-19-Impfung erfolgt war und bei denen keine zurückliegende SARS-CoV-2-Infektion bekannt war. Die fehlende Immunität wurde beim Screening durch einen negativen SARS-CoV-2-Antikörpertest gesichert. Nach Randomisierung erfolgte außerdem ein PCR-Test auf SARS-CoV-2, der bei 0,5 % (n = 25) der Studienteilnehmer positiv ausfiel.
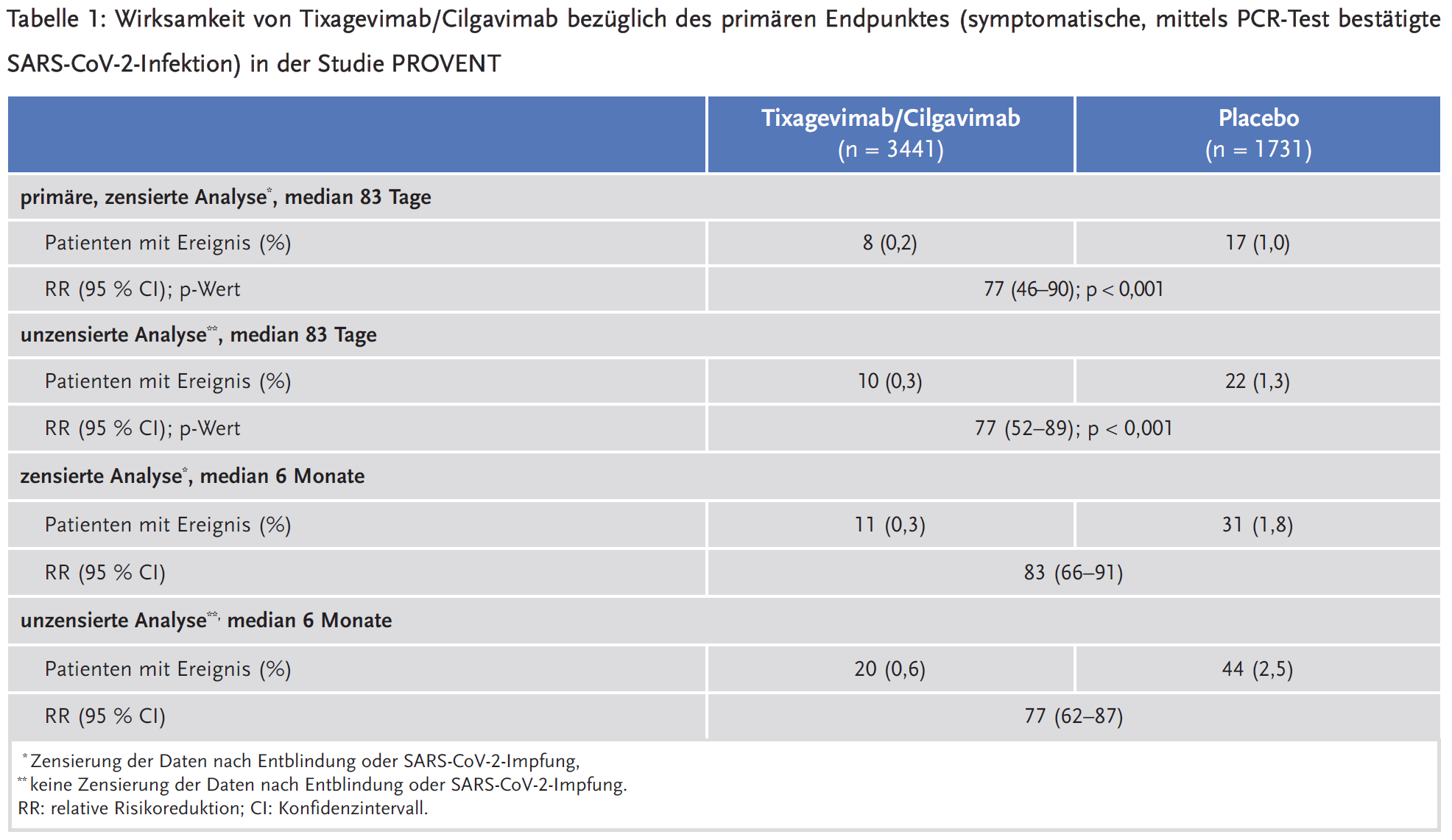
Primärer Endpunkt der Studie war die Häufigkeit symptomatischer, mittels PCR-Test bestätigter SARS-CoV-2-Infektionen. Als „symptomatisch“ galt jedes Anzeichen einer respiratorischen oder gastrointestinalen Infektion, das mindestens zwei Tage anhielt (z. B. Halsschmerzen, Schnupfen, Husten, Durchfall, Übelkeit) oder – unabhängig von der Dauer – Symptome einer schweren Infektion wie Fieber oder Atemnot. Aufgrund von Protokolländerungen erfolgte die primäre Analyse, als 30 % der Teilnehmer entblindet waren. Die Entblindung geschah fast ausschließlich (> 99 %) auf Wunsch der Patienten, die diese Information in ihre Impfentscheidung einbeziehen wollten. Patienten, die entblindet waren oder im Verlauf der Studie eine Impfung gegen COVID-19 erhalten hatten, wurden in der primären Analyse zensiert. Die mediane Beobachtungszeit bis zur primären Analyse betrug 83 Tage. Zu diesem Zeitpunkt waren insgesamt 25 symptomatische, mittels PCR-Test bestätigte SARS-CoV-2-Infektionen aufgetreten, davon 8 unter Tixagevimab/Cilgavimab und 17 unter Placebo. Dies entspricht einer signifikanten relativen Risikoreduktion von 77 % und einer absoluten Risikoreduktion von 0,8 %.
In einer ergänzenden Analyse erfolgte keine Zensierung: Alle randomisierten Patienten, die bei Studienbeginn einen negativen PCR-Test auf SARS-CoV-2 hatten, wurden einbezogen, auch wenn sie zwischenzeitlich entblindet oder gegen COVID-19 geimpft worden waren. Die unzensierten Daten ergaben bei einem Follow-up von 83 Tagen die gleichen Ergebnisse wie die primäre, zensierte Analyse. Im Median sechs Monate nach Randomisierung erfolgte eine weitere, im ursprünglichen Protokoll nicht vorgesehene Auswertung. Hierbei waren 47 % (Tixagevimab/Cilgavimab) bzw. 43 % (Placebo) der Patienten entblindet oder gegen COVID-19 geimpft. Die 6-Monats-Auswertung ergab im Vergleich zur primären Analyse eine etwas höhere relative Risikoreduktion (RR), wenn Patienten nach Entblindung oder Impfung zensiert wurden (RR 83 %). Bei Verzicht auf die Zensierung lag die RR nach sechs Monaten konstant bei 77 % (siehe Tabelle 1). Da nicht anzunehmen ist, dass in der Versorgung vor der Gabe von Tixagevimab/Cilgavimab immer eine akute SARS-CoV-2-Infektion mittels PCR-Test ausgeschlossen wird, schloss eine explorative Analyse auch Patienten ein, die bei Studienbeginn einen positiven PCR-Test aufwiesen. Die Wirksamkeit von Tixagevimab/Cilgavimab war in dieser Analyse weitgehend unverändert (RR 78; 95 % Konfidenzintervall [CI] 59–88).
Ein sekundärer Endpunkt der Studie untersuchte Serokonversionen gegenüber SARS-CoV-2. Bis Tag 83 hatten 1,3 % der Patienten unter Placebo und 0,7 % der Patienten unter Tixagevimab/Cilgavimab neu nachgewiesene Antikörper gegenüber SARS-CoV-2, nach sechs Monaten waren es 2,7 % bzw. 1,2 %. Dies entspricht einer signifikanten relativen Risikoreduktion von 51 % nach 83 Tagen bzw. 58 % nach sechs Monaten. Asymptomatische Infektionen wurden somit in einem etwas geringeren Ausmaß durch Tixagevimab/Cilgavimab verhindert als symptomatische Infektionen.
Die Studie PROVENT ist nicht ausreichend gepowert, um Aussagen über die Verhinderung schwerer COVID-19-Verläufe zu treffen. Als „schwer“ erkrankt galten in der Zulassungsstudie Patienten, die wegen einer Pneumonie oder Hypoxie stationär aufgenommen wurden und sauerstoffpflichtig waren oder beatmet werden mussten (WHO Clinical Progression Scale ≥ 5) (8). Im Beobachtungszeitraum von sechs Monaten traten bei fünf Patienten im Placebo-Arm und keinem Patienten unter Tixagevimab/Cilgavimab schwere COVID-19-Verläufe entsprechend dieser Definition auf. Bei sechs Patienten unter Tixagevimab/Cilgavimab (vs. 0 unter Placebo) erfolgte eine Abklärung COVID-19-verdächtiger Symptome in der Rettungsstelle. Todesfälle waren in beiden Armen selten (Tixagevimab/Cilgavimab vs. Placebo: primäre Analyse 0,1 % vs. 0,2 %; Follow-up nach sechs Monaten: 0,3 % vs. 0,4 %). Zwei Todesfälle im Placebo-Arm wurden als COVID-19-bedingt eingeschätzt (vs. 0 unter Tixagevimab/Cilgavimab).
Aufgrund der raschen Entwicklung neuer SARS-CoV-2-Varianten haben viele bislang entwickelte monoklonalen Antikörper ihre Schutzwirkung verloren. Eine verringerte Wirksamkeit besteht insbesondere gegenüber den Omikron-Varianten, da diese mehr als 35 Mutationen im Spike-Protein aufweisen (7). Zum Zeitpunkt der Studiendurchführung von PROVENT (November 2020 bis März 2021) waren die Alpha-, Beta-, Gamma- und Delta-Varianten von SARS-CoV-2 vorherrschend. Die Studie PROVENT kann deshalb eine Wirksamkeit gegenüber Omikron nicht belegen. In Labortests wies Tixagevimab/Cilgavimab auch gegenüber den Omikron-Varianten eine neutralisierende Wirkung auf, die allerdings schwächer ausfiel als gegenüber den Alpha-, Beta-, Gamma- und Delta-Varianten. Die neutralisierende Wirkung von Tixagevimab/Cilgavimab war gegenüber der Subvariante BA.1 moderat, gegenüber der Subvariante BA.2 gering reduziert (9). Aktuelle Untersuchungen zeigen eine achtfach reduzierte serologische Wirksamkeit von Tixagevimab/Cilgavimab gegenüber der Subvariante BA.4/.5 im Vergleich zu BA.2 (10). Es ist derzeit unklar, wie die in vitro verminderte serologische Wirksamkeit mit klinischen Ergebnissen korreliert.
Ausgewählte Nebenwirkungen
In der Studie PROVENT traten innerhalb von sechs Monaten numerisch mehr schwerwiegende kardiovaskuläre Ereignisse unter Tixagevimab/Cilgavimab auf als unter Placebo (0,7 % vs. 0,3 %), darunter auch vier kardiovaskuläre Todesfälle (vs. 0 unter Placebo). Alle Patienten mit schwerwiegenden kardiovaskulären Ereignissen hatten kardiovaskuläre Risikofaktoren oder Vorerkrankungen.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Monoklonale Antikörper werden aufgrund ihres hohen Molekulargewichts nicht renal ausgeschieden. Ihr Abbau erfolgt durch Proteolyse auf zellulärer Ebene. Eine Dosisanpassung bei Patienten mit Nieren- oder Leberfunktionsstörungen ist deshalb nicht erforderlich. Auch Wechselwirkungen mit anderen Arzneimitteln sind nicht zu erwarten.
- Tixagevimab und Cilgavimab müssen als separate, aufeinanderfolgende intramuskuläre Injektionen verabreicht werden. Aus Sicht der AkdÄ ist die i.m.-Injektion von Tixagevimab/Cilgavimab bei antikoagulierten Patienten nur bei Hochrisikopatienten vertretbar und sollte dann analog den Empfehlungen des RKI zur SARS-CoV-2-Impfung erfolgen, d. h. mit sehr feiner Kanüle und anschließender Kompression der Einstichstelle über zwei Minuten (11). Zur besseren postinterventionellen Kompression kann alternativ auch der Musculus deltoideus als Injektionsort gewählt werden.
- In der Studie PROVENT trat eine Anaphylaxie unter Tixagevimab/Cilgavimab auf. Schwere Überempfindlichkeitsreaktionen sind bekannte, seltene Ereignisse unter monoklonalen Antikörpern (12). Die Anwendung von Tixagevimab/Cilgavimab sollte deshalb unter Bedingungen erfolgen, in denen eine Behandlung anaphylaktischer Reaktionen rasch erfolgen kann.
- Ein erhöhtes Risiko für kardiovaskuläre Ereignisse unter Tixagevimab/Cilgavimab kann aktuell nicht ausgeschlossen werden, sodass im Rahmen der Aufklärung darauf hinzuweisen ist.
Dosierung und Kosten

Weiterführende Informationen
Tixagevimab/Cilgavimab (Evusheld®) wurde bislang noch nicht in die Bewertung des Zusatznutzens nach § 35a SGB V vom G-BA aufgenommen.
Quelle: Europäischer Öffentlicher Beurteilungsbericht (EPAR) Evusheld®, erschienen am 28. April 2022. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Literatur
- Bundesministerium für Gesundheit (BMG): Bundesanzeiger: Dritte Verordnung zur Änderung der SARS-CoV-2-Arzneimittelversorgungsverordnung vom 25. Mai 2022: https://www.bundesanzeiger.de/pub/publication/kryp49hsf6EZUBt7S26/content/kryp49hsf6EZUBt7S26/BAnz%20AT%2030.05.2022%20V1.pdf?inline (letzter Zugriff: 23. Juni 2021). Berlin, 30. Mai 2022.
- Levin MJ, Ustianowski A, De Wit S et al.: Intramuscular AZD7442 (Tixagevimab-Cilgavimab) for Prevention of Covid-19. N Engl J Med 2022; 386: 2188-2200.
- Food and drug administration (FDA): Fact sheet for healthcare providers: emergency use authorization for Evusheld™ (tixagevimab co-packaged with cilgavimab): https://www.fda.gov/media/154701/download (letzter Zugriff: 23. Juni 2022). Stand: Juni 2022.
- Robert Koch-Institut (RKI): Epidemiologisches Bulletin 39/2021: STIKO-Empfehlung zur COVID-19-Impfung: Personen mit Immundefizienz - Koadministration mit Totimpfstoffen: https://www.rki.de/DE/Content/Infekt/EpidBull/Archiv/2021/Ausgaben/39_21.pdf?__blob=publicationFile (letzter Zugriff: 23. Juni 2022). Berlin, Stand: 30. September 2021.
- Voysey M, Clemens SAC, Madhi SA et al.: Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021; 397: 99-111.
- Feng S, Phillips DJ, White T et al.: Correlates of protection against symptomatic and asymptomatic SARS-CoV-2 infection. Nat Med 2021; 27: 2032-2040.
- Pantaleo G, Correia B, Fenwick C et al.: Antibodies to combat viral infections: development strategies and progress. Nat Rev Drug Discov 2022.
- WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection: A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis 2020; 20: e192-e197.
- European Medicines Agency (EMA): Evusheld® – Tixagevimab + Cilgavimab: EPAR (Product Information Report): https://www.ema.europa.eu/en/documents/product-information/evusheld-epar-product-information_de.pdf (letzter Zugriff: 23. Juni 2022). EMA/205600/2022, Procedure No. EMEA/H/C/005788/0000. Amsterdam, 24. März 2022, first published 30. März 2022.
- Tuekprakhon A, Huo J, Nutalai R, Dijokaite-Guraliuc A: Further antibody escape by Omicron BA.4 and BA.5 from vaccine and BA.1 serum: https://doi.org/10.1101/2022.05.21.492554 (letzter Zugriff: 23. Juni 2021). 23. Mai 2022.
- Robert Koch-Institut (RKI): Epidemiologisches Bulletin 2/2021: Beschluss der STIKO zur 1. Aktualisierung der COVID-19-Impfempfehlung: https://www.rki.de/DE/Content/Infekt/EpidBull/Archiv/2021/Ausgaben/02_21.pdf?__blob=publicationFile (letzter Zugriff: 23. Juni 2022). Berlin, Stand: 14. Januar 2021.
- Yu RJ, Krantz MS, Phillips EJ, Stone CA, Jr.: Emerging causes of drug-induced anaphylaxis: a review of anaphylaxis-associated reports in the FDA Adverse Event Reporting System (FAERS). J Allergy Clin Immunol Pract 2021; 9: 819-829, e812.