Melatonin (Slenyto®)
Zugelassene Indikation
Slenyto® (retardiertes Melatonin) ist indiziert für die Behandlung von Schlafstörungen (Insomnie) bei Kindern und Jugendlichen im Alter von 2–18 Jahren mit Autismus-Spektrum-Störung (ASS) und/oder Smith-Magenis-Syndrom (SMS), wenn Schlafhygienemaßnahmen unzureichend waren.
Markteinführung
Slenyto® (retardiertes Melatonin) erhielt die „positive Opinion“ des Committee for Medicinal Products for Human Use (CHMP) am 26. Juli 2018. Die deutsche Markteinführung war am 15. Januar 2019.
Bewertung
Slenyto® (retardiertes Melatonin) wurde im Rahmen einer „Paediatric Use Marketing Authorisation“ (PUMA) zugelassen. Der Wirkstoff ist bekannt und bereits für Erwachsene zugelassen. Die vorliegende Zulassung deckt einen „unmet clinical need“, da diesen Patienten bisher unretardiertes Melatonin off-label verschrieben wurde.
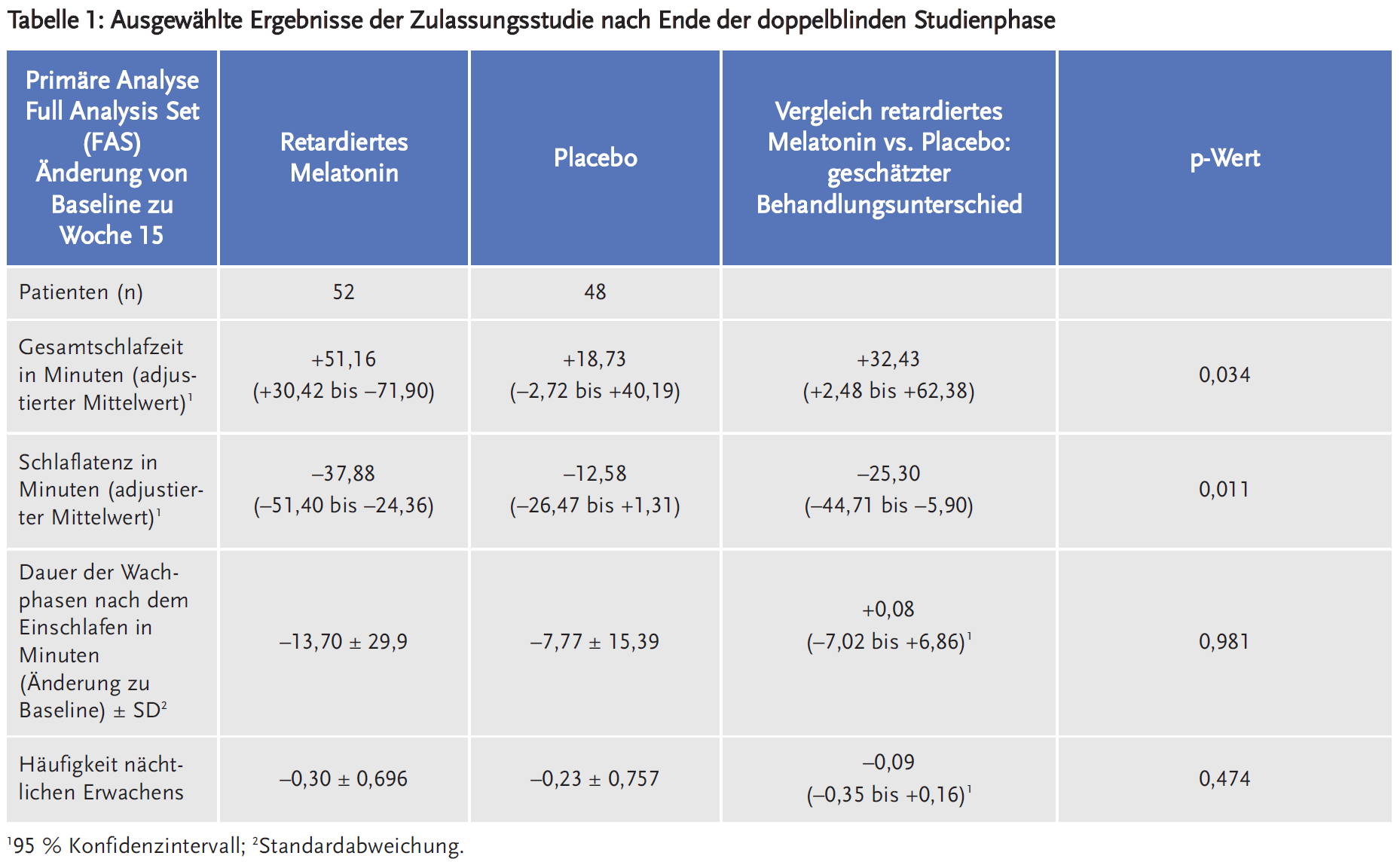
Wirksamkeit in den Zulassungsstudien
Die pivotale Studie ist eine multinationale, multizentrische, randomisierte, doppelblinde, placebokontrollierte Studie. Zunächst erfolgte eine einfach verblindete zweiwöchige Placebo-Run-in-Phase, auf welche die doppelblinde Wirksamkeits-/Sicherheitsphase von 13 Wochen folgte. Darauf folgten 13 Wochen Open-Label-Behandlung und danach weitere 78 Wochen Open-Label-Behandlung mit optionaler Dosiseskalation und schließlich eine zweiwöchige einfach verblindete Placebo-Run-out-Phase, um einen eventuellen Entzug zu beurteilen. Die Patienten bekamen initial 2 mg retardiertes Melatonin (rM) abends, mit der Option auf 5 mg oder auch 10 mg zu eskalieren (aber auch wieder zu reduzieren). Insgesamt lag die Studiendauer bei 2,2 Jahren.
Der primäre Endpunkt war die Gesamtschlafdauer. Sekundäre Endpunkte waren u. a. Schlaflatenz, Häufigkeit nächtlichen Erwachens und Dauer der Wachphasen nach dem Einschlafen.
Es wurden insgesamt 125 Kinder mit ASS (n = 121) oder SMS (n = 4) eingeschlossen. Das mittlere Alter war 8,7 (± Standardabweichung 4,15) Jahre, 73,6 % waren Jungen.
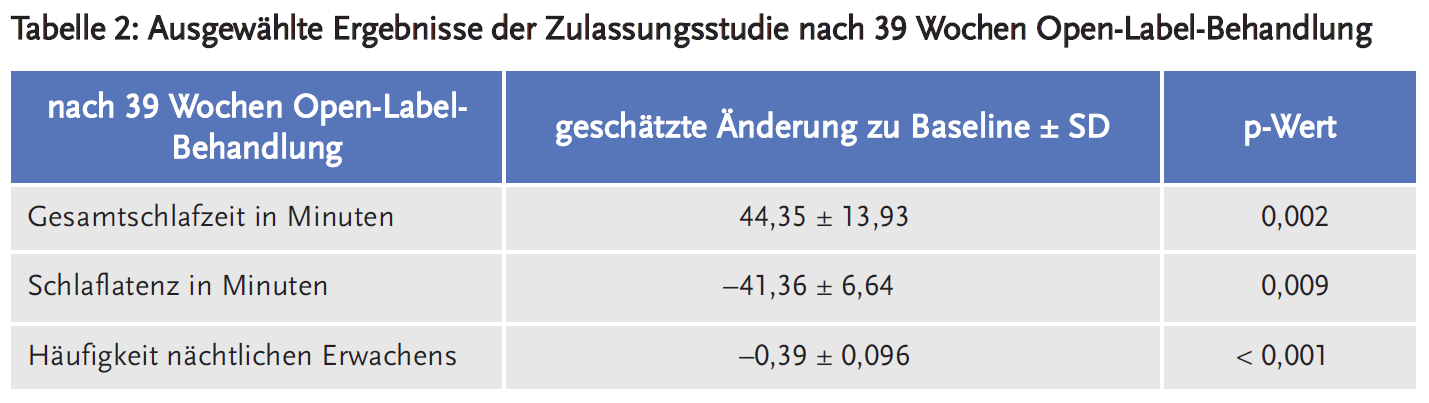
Es liegen weitere Daten für 79 Patienten vor, welche 39 Wochen eine Open-Label-Behandlung absolvierten. Hier stellten sich sowohl in der Gruppe der Patienten aus dem rM-Arm als auch dem Placebo-Arm ähnliche Veränderungen ein.
Ebenfalls verbesserte sich die Tagesmüdigkeit und Lebensqualität der Bezugspersonen.
Ausgewählte Nebenwirkungen
Etwa 84,2 % der Patienten erlitten Nebenwirkungen während der Studie. Diese waren jedoch insgesamt milder Ausprägung. Am häufigsten waren dies Erschöpfung, morgendliche Müdigkeit, Somnolenz, plötzliche Schlafattacken und Kopfschmerzen.
Die Langzeitsicherheit der Melatonin-Anwendung bei Kindern ist jedoch bisher nicht umfassend untersucht. Da es am Ende der Pubertät zu einem Abfall der endogenen Melatonin-Produktion kommt, könnte die exogene Melatonin-Zufuhr durch supraphysiologische Plasmakonzentrationen die Pubertät verzögern.
Fazit
Retardiertes Melatonin scheint aufgrund dieser Ergebnisse eine wirksame und sichere Therapie für pädiatrische Patienten mit ASS oder SMS und Schlafstörungen zu sein, bei denen Schlafhygienemaßnahmen nicht wirksam waren. Insbesondere stellt es eine zugelassene medikamentöse Therapie in altersgerechter Darreichungsform dar, sodass ein Off-Label-Use von anderen, eigentlich ungeeigneten Präparaten in dieser pädiatrischen Population endlich nicht mehr notwendig ist.
Die Kinder schlafen im Mittel 25 Minuten schneller ein und 32 Minuten länger. Für die Praxis ist es jedoch weiterhin wichtig, diejenigen Patienten zu identifizieren, welche nicht auf Melatonin ansprechen und entsprechend alternative Therapien einzuleiten.

Weiterführende Informationen
Das IQWiG wurde am 15. Januar 2019 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Slenyto®, erschienen am 10. Oktober 2018. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 29. März 2019 vorab online veröffentlicht.