Galcanezumab (Emgality®) ▼
Zugelassene Indikation und Wirkmechanismus
Galcanezumab (Emgality®) ist als subkutane Injektion zur Migräneprophylaxe bei Erwachsenen mit mindestens vier Migränetagen pro Monat zugelassen.
Galcanezumab ist ein rekombinanter humanisierter monoklonaler IgG4-Antikörper, der in Ovarialzellen chinesischer Hamster (CHO) hergestellt wird. Er bindet an das Calcitonin-Gene-Related-Peptide (CGRP) und hindert dieses somit an der Aktivierung seines Rezeptors, der in der Pathophysiologie der Migräne eine zentrale Rolle spielt. CGRP ist ein Neuropeptid, das die nozizeptive Signalübertragung reguliert und als Vasodilatator wirkt. Der CGRP-Spiegel steigt während eines Migräneanfalls an und normalisiert sich beim Abklingen der Kopfschmerzen. Galcanezumab konkurriert spezifisch mit CGRP um die Bindung am CGRP-Rezeptor und hemmt dadurch seine Funktion.
Galcanezumab wird subkutan am Abdomen, am Oberschenkel, an der Außenseite des Oberarms oder in den Gesäßbereich appliziert. Patienten sollen nach angemessener Schulung Galcanezumab selbst verabreichen. Die empfohlene Dosis beträgt 120 mg Galcanezumab einmal monatlich, die Behandlung wird mit einer Initialdosis von 240 mg (zwei Injektionen zu je 120 mg am selben Tag) eingeleitet. Galcanezumab muss im Kühlschrank bei 2–8°C gelagert werden. Ungekühlt kann das Arzneimittel bis zu sieben Tage bei bis zu 30 °C gelagert werden.
Markteinführung
Galcanezumab (Emgality®) ist seit dem 1. April 2019 in der o. g. Indikation auf dem deutschen Markt.
Bewertung
Galcanezumab ist der zweite monoklonale Antikörper, der sich spezifisch gegen das Migräne auslösende Neuropeptid Calcitonin-Gene-Related-Peptide (CGRP) richtet. Galcanezumab reduziert die durchschnittlichen Migränetage um 4,2–4,7 Tage pro Monat (bei episodischer Migräne, EM) und um 4,6–4,8 Tage pro Monat (bei chronischer Migräne, CM). Der Unterschied zu Placebo (Reduktion um 2,3–2,8 Tage bei EM und um 2,7 Tage bei CM) ist statistisch signifikant. Unter Galcanezumab wird eine mindestens 50-prozentige Reduktion der monatlichen Migränetage bei etwa 28 % der Patienten mit CM (versus 15 % unter Placebo; NNT = 8) sowie bei 57–62 % der Patienten mit EM (versus 36–37 % unter Placebo; NNT = 4) berichtet. Auch Begleitsymptome wie Übelkeit, Erbrechen oder Lichtempfindlichkeit sollen durch Galcanezumab im Vergleich zu Placebo statistisch signifikant verbessert werden. Galcanezumab verringerte statistisch signifikant besser als Placebo die Anzahl der Tage, an denen eine akute Migränemedikation erforderlich war: bei CM im Mittel um 4,7 (120 mg) bzw. 4,3 (240 mg) Tage versus 2,2 Tage unter Placebo, bei EM im Mittel um 3,7–4 (120 mg) bzw. 3,7 (240 mg) Tage versus 2 Tage unter Placebo.
Unerwünschte Arzneimittelwirkungen wie Schmerzen (10,1 % unter 120 mg, 11,6 % unter 240 mg) und Reaktionen an der Injektionsstelle (9,9 % bzw. 14,5 %) traten sehr häufig auf. Vertigo, Obstipation und Pruritus waren unter Galcanezumab häufiger als unter Placebo.
Galcanezumab bietet – wie bereits der erste CGRP-Rezeptorantagonist Erenumab1 – gegenüber den verfügbaren Alternativen zur Migräneprophylaxe einen vergleichbaren Effekt. Der Vorteil gegenüber bisher verfügbaren Wirkstoffen scheint in der besseren Verträglichkeit zu liegen. Ein weiterer Vorteil könnte die vierwöchentliche Applikation sein, die allerdings subkutan erfolgen muss. Es gibt keinen direkten Vergleich der beiden verfügbaren CGRP-Rezeptorantagonisten Erenumab und Galcanezumab.
CGRP hat eine ausgeprägte vasodilatatorische Wirkung. Die Hemmung seines Rezeptors birgt daher theoretisch das Risiko für kardiovaskuläre Ereignisse, das bei Migräne ohnehin gering erhöht ist. Die verfügbaren Studien ergaben keine eindeutigen Hinweise auf ein erhöhtes kardiovaskuläres Risiko, allerdings wurden Patienten > 65 Jahre oder mit kardiovaskulären Ereignissen in der Vorgeschichte ausgeschlossen. Die Risiken einer langfristigen Blockade von CGRP mit Galcanezumab – insbesondere hinsichtlich kardiovaskulärer Nebenwirkungen – können zum jetzigen Zeitpunkt nicht abschließend beurteilt werden, da Langzeitdaten zur Anwendung von Galcanezumab fehlen (kein Patient hat in den Zulassungsstudien das Arzneimittel länger als zwölf Monate bekommen). Der Einsatz von Galcanezumab sollte daher vorerst nur nach Versagen anderer Arzneimittel zur Migräneprophylaxe oder bei Unverträglichkeit erfolgen.
Wirksamkeit in den Zulassungsstudien
Galcanezumab wurde in zwei Zulassungsstudien bei episodischer Migräne (EVOLVE-1 und EVOLVE-2) sowie in einer Zulassungsstudie bei chronischer Migräne (REGAIN) untersucht.
Als primärer Endpunkt wurde die durchschnittliche Änderung der Anzahl der Migränetage pro Monat erhoben. Sekundäre Endpunkte waren u. a. die Ansprechrate (der Anteil der Patienten, die eine mindestens 50-, 75- und 100-prozentige Reduktion der Migränetage pro Monat erreichten) sowie die durchschnittliche Änderung der Anzahl der Tage mit akuter Migränemedikation im Vergleich zum Studienbeginn. Zudem wurden patientenrelevante Endpunkte wie der Einfluss auf die Beeinträchtigung der täglichen Aktivität (die durchschnittliche Änderung des Role Function-Restrictive-Score im Migraine-Specific Quality of Life Questionnaire (MSQ) sowie im Migraine Disability Assessment (MIDAS)) und die Patienteneinschätzung der subjektiven Symptomschwere erhoben (Patient Global Impression of Severity (PGI-S).
Die internationalen, multizentrischen, doppelblinden, randomisierten, placebokontrollierten Phase-III-Studien EVOLVE-1 und -2 schlossen Patienten mit episodischer Migräne (EM) mit oder ohne Aura ein, die in den vorausgegangenen drei Monaten 4–14 Migränetage pro Monat und mindestens zwei Migräneattacken hatten. Die Ausgangsbefunde wurden mittels eines elektronischen Tagebuchs über 30–40 Tage erhoben, daran schloss sich eine Behandlungsphase von sechs Monaten an. Die Patienten wurden im Verhältnis 2:1:1 randomisiert und erhielten subkutan alle vier Wochen Placebo, 120 mg Galcanezumab (nach Initialdosis von 240 mg) oder 240 mg Galcanezumab, stratifiziert nach geografischer Region und Häufigkeit der Migränetage pro Monat.
In die EVOLVE-1 wurden 862 Patienten randomisiert, nur 858 davon bekamen mindestens eine Dosis der Studienmedikation: Placebo (n = 433), 120 mg Galcanezumab (n = 213) oder 240 mg (n = 212) Galcanezumab. Die Patienten waren in den Galcanezumab-Armen im Median 40 Jahre alt und im Placebo-Arm im Median 41 Jahre alt und zu 84 % weiblich. Die mittlere Migränehäufigkeit bei Studienbeginn betrug etwa 9 Migränetage pro Monat, an im Mittel 7 davon erfolgte die Einnahme von migränespezifischer Medikation. 81,9 % der Patienten schlossen die doppelblinde Behandlungsphase ab (351 im Placebo-Arm, 177 im 120-mg-Arm und 175 im 240-mg-Arm).
In die EVOLVE-2 wurden 922 Patienten randomisiert, nur 915 davon bekamen mindestens eine Dosis der Studienmedikation: Placebo (n = 461), 120 mg Galcanezumab (n = 231) oder 240 mg (n = 223) Galcanezumab. Die Patienten waren in den Galcanezumab-Armen im Median 41 Jahre alt und im Placebo-Arm im Median 42 Jahre alt und zu 85 % weiblich. Die mittlere Migränehäufigkeit bei Studienbeginn betrug etwa 9 Migränetage pro Monat, an im Mittel 7,5 davon erfolgte die Einnahme von migränespezifischer Medikation. 85,8 % der Patienten schlossen die doppelblinde Behandlungsphase ab (387 im Placebo-Arm, 203 im 120-mg-Arm und 195 im 240-mg-Arm).
In die internationale, multizentrische, doppelblinde, randomisierte, placebokontrollierte REGAIN-Studie wurden Patienten mit chronischer Migräne (CM) mit oder ohne Aura und mit ≥ 15 Kopfschmerztage/Monat und davon ≥ 8 Migränetage/Monat in den vorausgegangenen drei Monaten eingeschlossen. Sie wurden im Verhältnis 2:1:1 randomisiert und erhielten subkutan alle vier Wochen Placebo (n = 558), 120 mg Galcanezumab (n = 278; nach Initialdosis von 240 mg) oder 240 mg Galcanezumab (n = 277), stratifiziert nach geografischer Region und Häufigkeit der Migränetage pro Monat. Die Patienten waren in den Galcanezumab-Armen im Median 40 Jahre alt und im Placebo-Arm im Median 41 Jahre alt und zu 83 % bzw. 87 % weiblich. Die mittlere Migränehäufigkeit bei Studienbeginn betrug etwa 19 Migränetage pro Monat, an im Mittel 15 davon erfolgte die Einnahme von migränespezifischer Medikation. Bei 63,8 % der Patienten wurde ein Übergebrauch von Akutmedikation berichtet. 77,8 % der Patienten hatten bereits mindestens eine medikamentöse Migräneprophylaxe, bei 29,5 % davon hatten bereits mindestens zwei Behandlungsoptionen versagt. Nach einer Beobachtungsphase von 30–40 Tagen, in der die Ausgangsbefunde erhoben wurden, erfolgte die Behandlungsphase über zwölf Wochen.
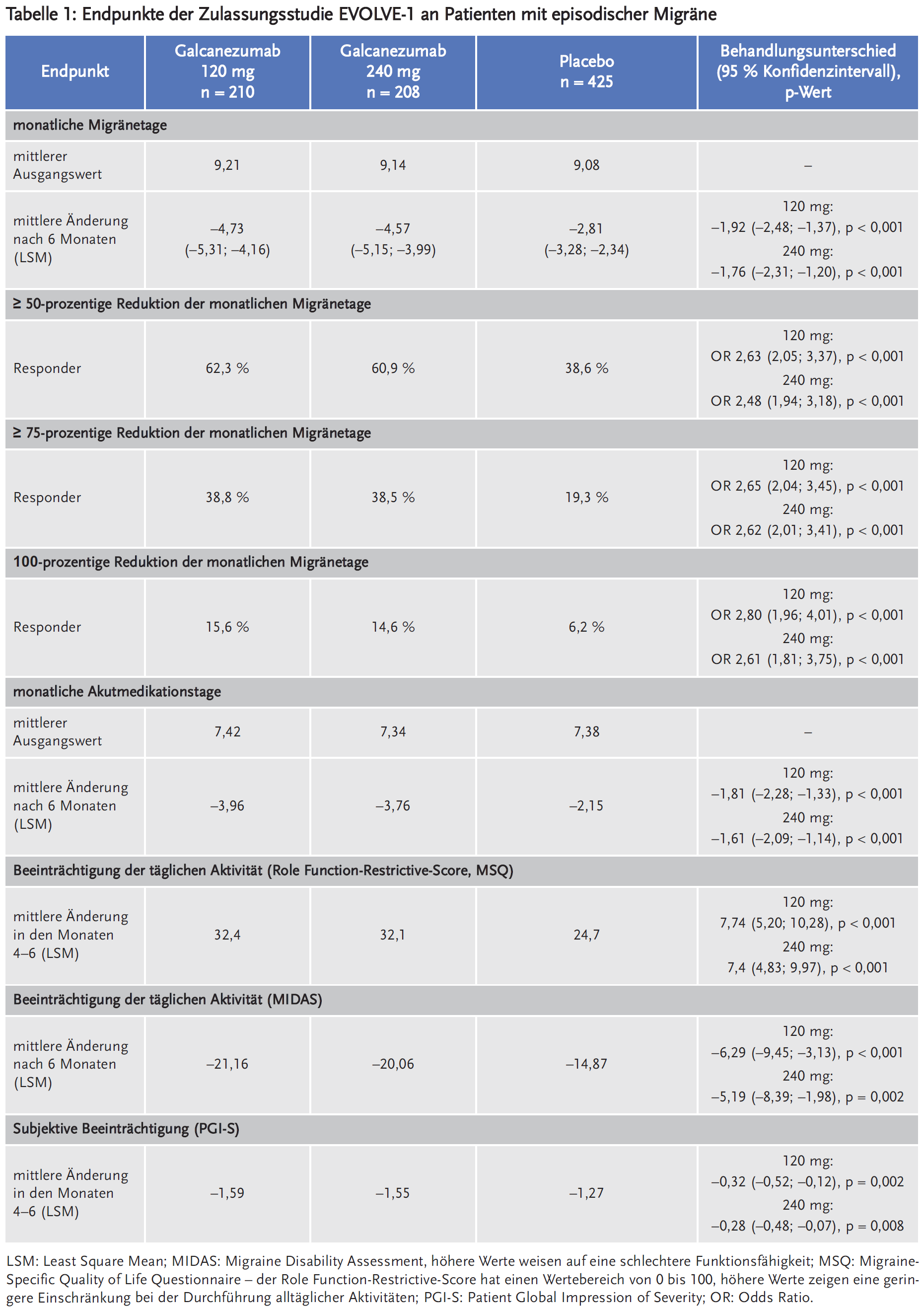
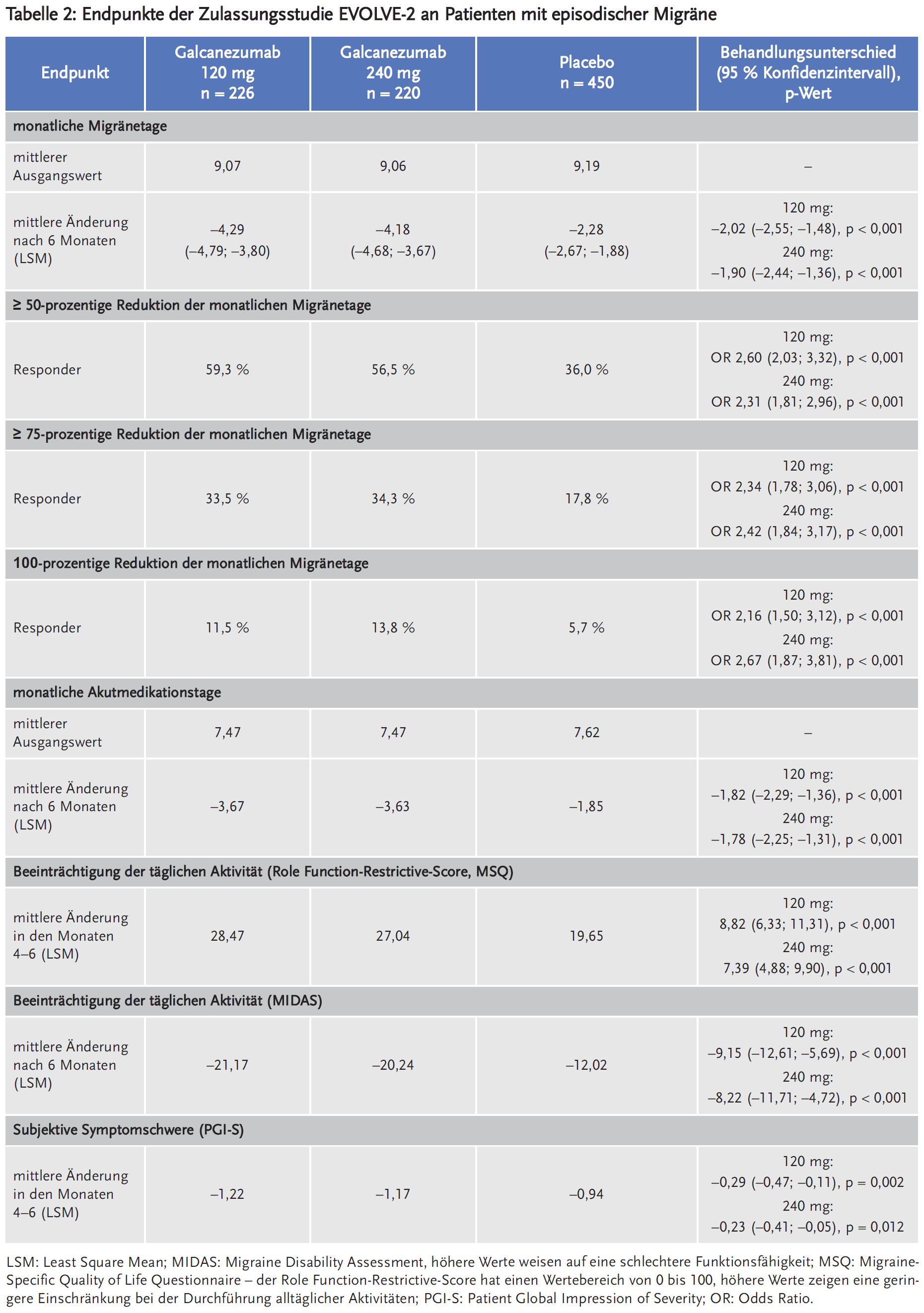
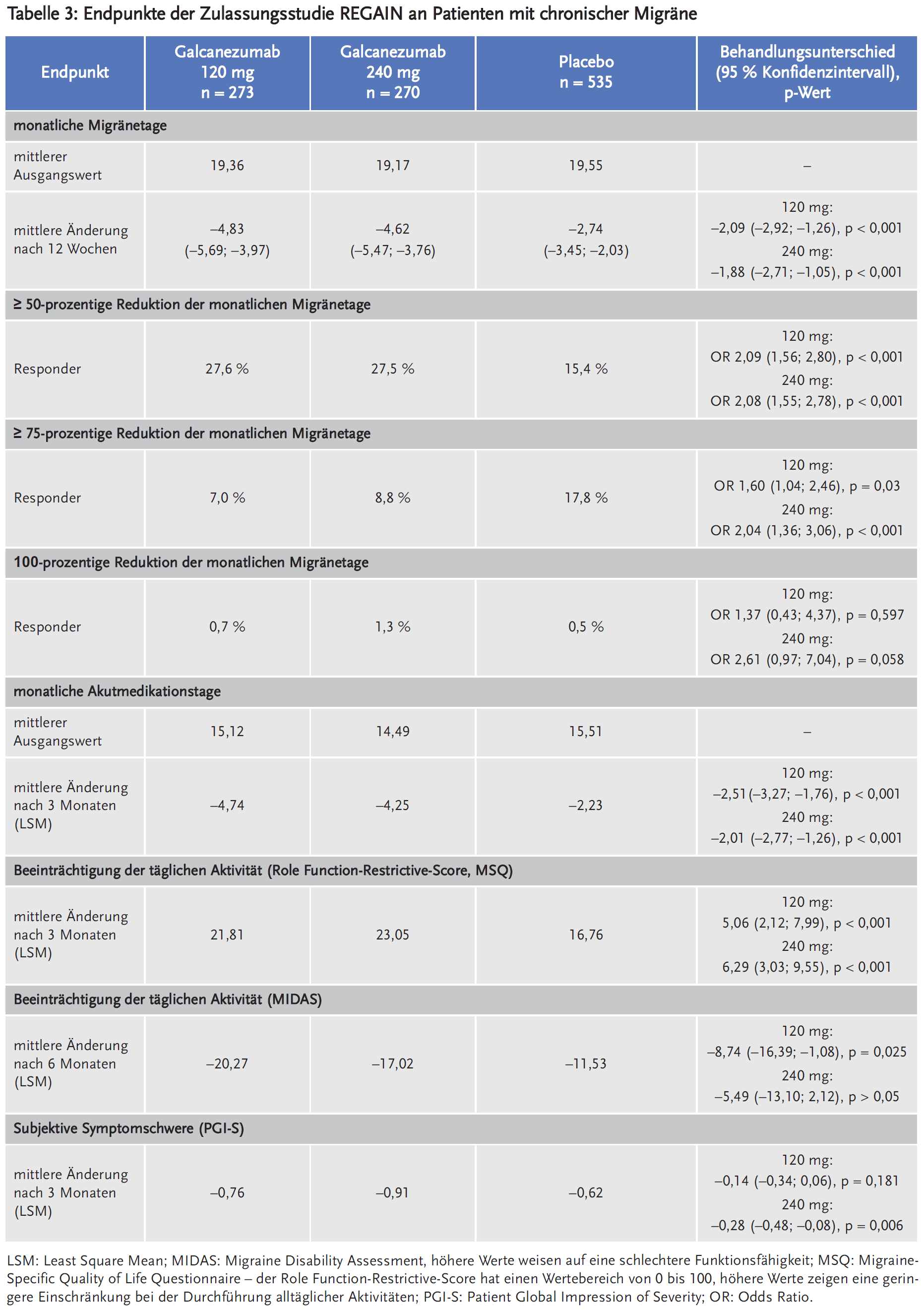
In allen drei Zulassungsstudien zeigten sich signifikante Unterschiede unter beiden Dosierungen von Galcanezumab im Vergleich zu Placebo bezüglich patientenrelevanter Wirksamkeitsendpunkte wie u. a. Reduktion der monatlichen Akutmedikationstage, der Ansprechraten sowie der Beeinträchtigung der täglichen Aktivität und der subjektiven Symptomschwere (Tabellen 1 bis 3). Bei EM reduzierte Galcanezumab die Anzahl der durchschnittlichen monatlichen Migränetage um 4,3 – 4,7 Tage (120 mg) bzw. 4,6 Tage (240 mg). Der Unterschied zu Placebo war statistisch signifikant. Zudem erreichte mehr als die Hälfte der Patienten unter Galcanezumab eine mindestens 50-prozentige Reduktion der monatlichen Migränetage. Dies entspricht einer Number needed to treat (NNT) von 4. Bei CM reduzierte Galcanezumab die Anzahl der durchschnittlichen monatlichen Migränetage um 4,8 Tage (120 mg) bzw. 4,6 Tage (240 mg). Der Unterschied zu Placebo war statistisch signifikant. Unter Galcanezumab erreichte nur etwa ein Viertel der Patienten eine mindestens 50-prozentige Reduktion der monatlichen Migränetage. Dies entspricht einer NNT von 8.
Zwischen den zwei evaluierten Dosierungen zeigten sich keine statistisch signifikanten Unterschiede, die höhere Dosierung von 240 mg zeigte in keinem Endpunkt erkennbar bessere Wirksamkeit als die 120-mg-Dosis.
Ausgewählte Nebenwirkungen
Schmerzen (10,1 % unter 120 mg, 11,6 % unter 240 mg) und Reaktionen an der Injektionsstelle (9,9 % bzw. 14,5 %) traten sehr häufig auf. Die wichtigsten häufigen Nebenwirkungen unter 120 mg waren Vertigo (0,7 %), Obstipation (1,0 %) und Pruritus (0,7 %) und Urtikaria (0,3 %).
In den Zulassungsstudien wurden schwerwiegende kardiovaskuläre unerwünschte Arzneimittelereignisse bei insgesamt sieben Patienten berichtet: akute Myokardinfarkte, Lungenembolie, TIA und instabile Angina pectoris.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
Galcanezumab könnte einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen haben. Nach der Verabreichung kann Vertigo auftreten.
Von der Teilnahme an den klinischen Studien waren ausgeschlossen: ältere Patienten (> 65 Jahre), Patienten mit vorbestehendem Myokardinfarkt, Schlaganfall, tiefen Venenthrombosen, transitorischen ischämischen Attacken, instabiler Angina pectoris, koronarer Bypass-Operation oder anderen durchgeführten Revaskularisierungsverfahren innerhalb der letzten sechs Monaten vor dem Screening sowie Patienten mit einem schwerwiegenden kardiovaskulären Risiko. Für diese Patientengruppen liegen weder Wirksamkeits- noch Sicherheitsdaten vor.

Weiterführende Informationen
Das IQWiG wurde am 01.04.2019 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Emgality®, erschienen am 14. Februar 2019. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Fußnote
1Vgl. Neue Arzneimittel Erenumab vom 14.12.2018: www.akdae.de/Arzneimitteltherapie/NA/Archiv-INN/201808-Aimovig.pdf.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 2. Mai 2019 vorab online veröffentlicht.