Rituximab (MabThera®), neue Indikation: Pemphigus vulgaris
Zugelassene Indikation und Wirkmechanismus
Rituximab (MabThera®) wurde bereits 1998 in der EU zur Behandlung von u. a. Non-Hodgkin-Lymphom, chronischer lymphatischer Leukämie und rheumatoider Arthritis zugelassen. Im März 2019 erfolgte eine Indikationserweiterung zur Behandlung von Patienten mit mäßigem bis schwerem Pemphigus vulgaris (PV).
PV gehört zu den blasenbildenden Autoimmundermatosen und ist eine seltene Erkrankung mit einer Inzidenz von etwa 0,5 bis 2 Neuerkrankungen pro 100.000 Einwohner jährlich. Charakteristisch für PV sind Autoantikörper gegen desmosomale Proteine der Epidermis (Desmogleine), die die Zerstörung der Desmosomen und eine daraus resultierende Akantholyse und makroskopische Blasenbildung bedingen.
Rituximab ist ein gentechnisch hergestellter monoklonaler chimärer Antikörper (Maus/Mensch). Es bindet spezifisch an das Transmembran-Antigen CD20, das auf Prä-B- und reifen B-Lymphozyten exprimiert wird. Rituximab bewirkt immunologische Reaktionen wie komplementabhängige Zytotoxizität und antikörperabhängige zelluläre Zytotoxizität, die eine B-Zell-Lyse initiieren.
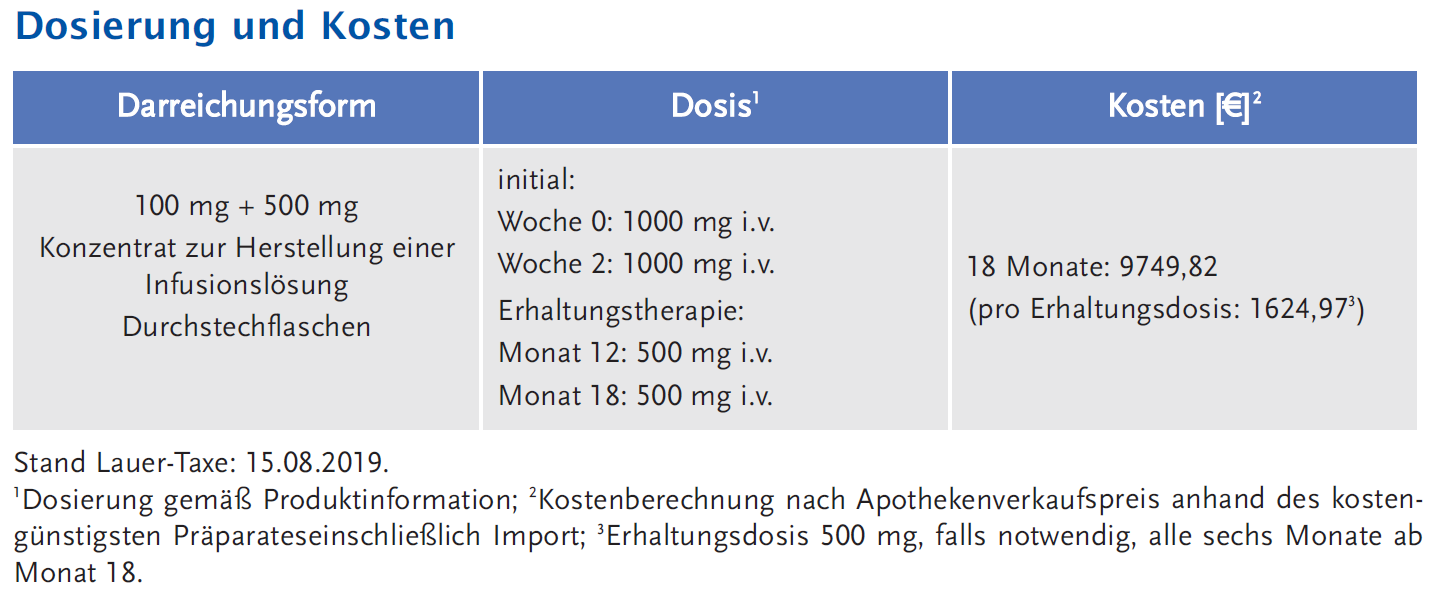
Die empfohlene Dosierung zur Behandlung von PV beträgt 1000 mg Rituximab als i.v. Infusion, gefolgt von einer zweiten i.v. Infusion von 1000 mg zwei Wochen später, in Kombination mit einem ausschleichenden Glukokortikoid-Zyklus. Als Erhaltungstherapie werden 500 mg Rituximab in den Monaten 12 und 18 als i.v. Infusion verabreicht, die – wenn klinisch erforderlich – alle 6 Monate wiederholt wird. Zur Behandlung eines Rückfalls können Patienten 1000 mg i.v. erhalten, nachfolgende Infusionen dürfen frühestens 16 Wochen nach der vorhergehenden Infusion verabreicht werden.
Markteinführung
Rituximab (MabThera®) ist seit 1998 auf dem deutschen Markt.
Bewertung
Im Vergleich zur Behandlung mit der Standarddosis von oralem Prednison erreichten unter Rituximab in Kombination mit niedrig dosiertem Prednison statistisch signifikant mehr Patienten mit mäßigem bis schwerem Pemphigus vulgaris nach 24 Monaten eine Komplettremission ohne Kortikosteroidtherapie für eine Dauer von zwei oder mehr Monaten. Numerische Unterschiede zwischen den Behandlungsarmen zugunsten von Rituximab zeigten sich auch bezüglich der Komplettremission unter minimaler Kortikosteroidtherapie sowie der Dauer der Komplettremission bei Patienten mit Ansprechen nach 24 Monaten. Zudem erlitten weniger Patienten unter Rituximab schwere oder mittelschwere Rückfälle. Durch die Gabe von Rituximab konnte während der 24-monatigen Behandlungsdauer ein steroidsparender Effekt erzielt werden.
In der Zulassungsstudie zeigten sich keine neuen oder unbekannten Nebenwirkungen unter der Behandlung mit Rituximab. Am häufigsten traten infusionsbedingte Reaktionen wie Kopfschmerzen, Schüttelfrost, hoher Blutdruck, Übelkeit, Asthenie und Schmerzen auf. 37 % der Patienten unter Rituximab hatten behandlungsbedingte Infektionen im Vergleich zu 42 % der Patienten im Prednison-Arm. Die häufigsten Infektionen unter Rituximab waren Herpes-zoster- und -simplex-Infektionen, Bronchitis, Harnwegsinfektion, Pilzinfektion und Konjunktivitis.
Damit erscheint das Risiko-Nutzen-Profil von Rituximab bei mäßigem bis schwerem Pemphigus vulgaris als positiv. Mit einer NNT von 1,6 im Vergleich zu Prednison allein bezüglich der Komplettremission (89,5 % versus 27,3 %) eröffnet Rituximab eine neue, wirksame Behandlungsmöglichkeit für diese seltene, schwere Erkrankung. Ob Rituximab auch im Vergleich zu anderen Behandlungsoptionen wie Azathioprin, Mycophenolatmofetil, Cyclophosphamid und Methotrexat vergleichbare Effekte zeigt, ist unklar. Zudem lässt sich anhand der vorliegenden Daten nicht abschließend beurteilen, ob die Behandlung mit Rituximab die gesundheitsbezogene Lebensqualität der Patienten positiv beeinflusst.
Wirksamkeit in den Zulassungsstudien
Die Wirksamkeit und Sicherheit von Rituximab in Kombination mit einer kurzzeitigen, niedrig dosierten Behandlung mit oralem Prednison wurde im Vergleich zu einer Langzeitbehandlung mit oralem Prednison in einer multizentrischen, offenen, randomisierten, kontrollierten Phase-III-Studie (Ritux 3) untersucht. Die Studie schloss 90 erwachsene Patienten mit neu diagnostiziertem, mäßigem bis schwerem Pemphigus (74 mit PV, 16 mit Pemphigus foliaceus, PF) ein, die stratifiziert nach dem Schweregrad der Erkrankung bei Baseline entweder Rituximab + Prednison (Rituximab-Arm, n = 46) oder Prednison (Prednison-Arm, n = 44) für 24 Monate bekamen. Die Dosierung von Prednison betrug im Rituximab-Arm 0,5 oder 1 mg Prednison pro kg Körpergewicht (KG) und Tag für 3 oder 6 Monate je nach Schweregrad und im Prednison-Arm 1 oder 1,5 mg Prednison pro kg KG und Tag für 12 oder 18 Monate je nach Schweregrad.
Als primärer Endpunkt wurde die Komplettremission – vollständige Epithelialisierung und Abwesenheit neuer und/oder bestehender Läsionen – ohne Kortikosteroidtherapie für eine Dauer von 2 oder mehr Monaten nach 24 Monaten erhoben. Sekundäre Endpunkte waren u. a. Komplettremission unter minimaler Kortikosteroidtherapie nach 24 Monaten, die Anzahl der Patienten mit mindestens einem schweren oder mittelschweren Rückfall nach 24 Monaten, die Dauer der Komplettremission ohne Kortikosteroidtherapie nach 24 Monaten bei Patienten mit Ansprechen, die kumulative Dosis an Prednison nach 24 Monaten und die gesundheitsbezogene Lebensqualität.
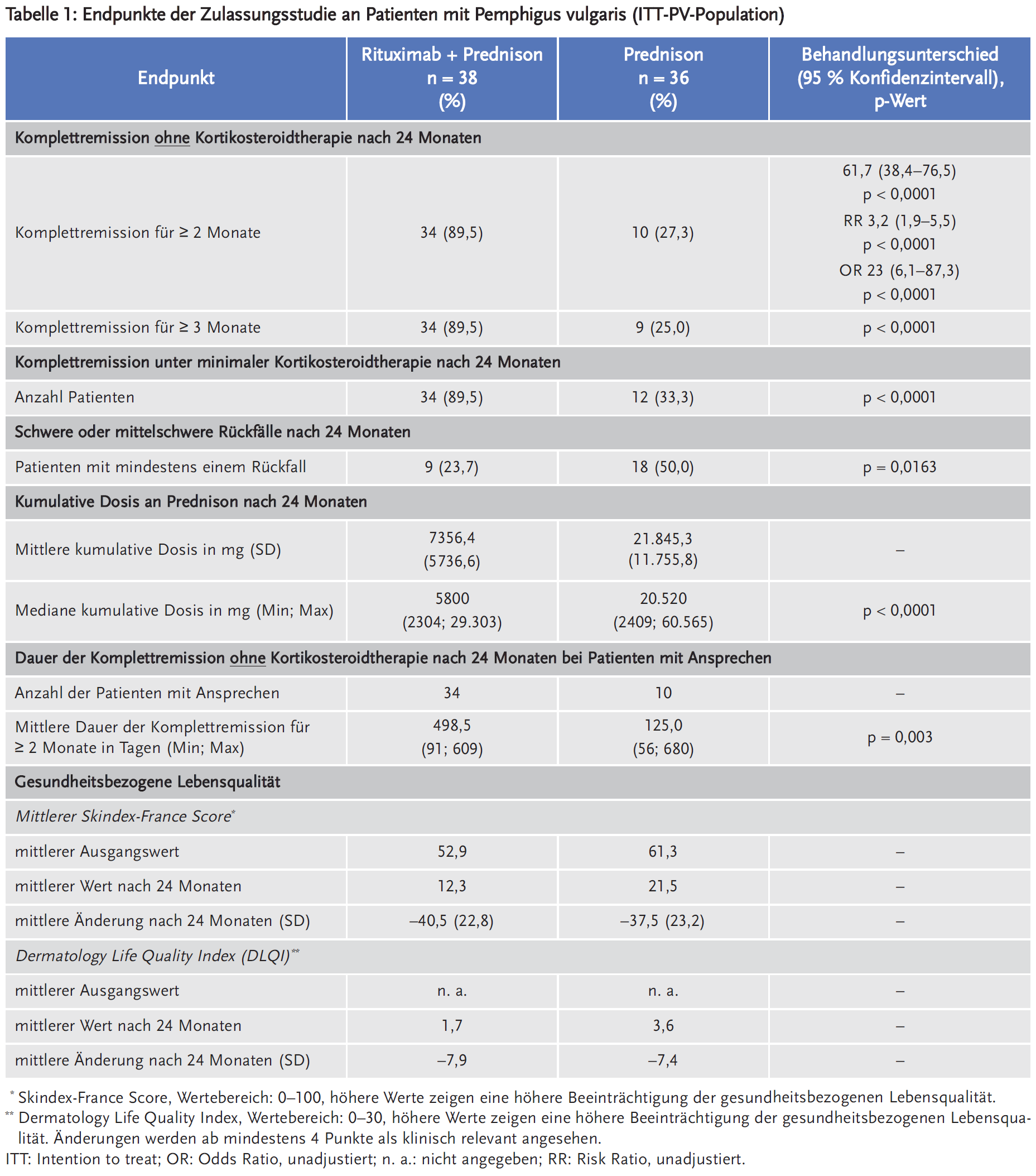
Unter Rituximab + Prednison erreichten statistisch signifikant mehr Patienten eine Komplettremission ohne Kortikosteroidtherapie in Monat 24 für eine Dauer von zwei oder mehr Monaten. Numerische Unterschiede zwischen den Behandlungsarmen zeigten sich auch bezüglich der Komplettremission unter minimaler Kortikosteroidtherapie nach 24 Monaten sowie der Dauer der Komplettremission bei Patienten mit Ansprechen. Weniger Patienten erlitten unter der Gabe von Rituximab schwere oder mittelschwere Rückfälle nach 24 Monaten sowie in der Nachbeobachtungsphase von Monat 24 bis zu Monat 36 (2 Patienten unter Rituximab + Prednison vs. 11 Patienten unter Prednison). Im Monat 36 konnten 94 % (32/34) der Patienten im Rituximab-Arm und 60 % (6/10) der Patienten im Prednison-Arm die erreichte Komplettremission aufrechterhalten, wobei die Untersuchung im Monat 36 erst später ins Studienprotokoll aufgenommen wurde, als einige Patienten diesen Zeitpunkt bereits überschritten hatten. Daten zu Monat 36 wurden daher nur für 71 % zum genauen Zeitpunkt sowie für 22 % post hoc erhoben.
Der Effekt auf die gesundheitsbezogene Lebensqualität war in beiden Behandlungsarmen vergleichbar, sodass kein Vorteil für Rituximab gezeigt werden konnte.
Studienabbrüche kamen bei 2 (4 %) der Patienten im Rituximab-Arm und 13 (30 %) der Patienten im Prednison-Arm vor, wobei insbesondere im Prednison-Arm die häufigsten Ursachen dafür Nebenwirkungen (n = 8) und fehlende Wirksamkeit bzw. Rückfälle (n = 4) waren. Im Rituximab-Arm brach je ein Patient die Studie aufgrund von Nebenwirkungen bzw. fehlender Krankheitskontrolle vorzeitig ab.
Derzeit läuft noch die multizentrische, randomisierte, doppelblinde PEMPHIX-Studie, die Rituximab mit Mycophenolatmofetil bei 135 Patienten mit mäßigem bis schwerem PV vergleicht. Ergebnisse sollen im November 2019 vorliegen1 .
Ausgewählte Nebenwirkungen
In der Studie traten im Rituximab-Arm keine neuen oder unbekannten Nebenwirkungen auf. 58 % der Patienten in diesem Arm hatten infusionsbedingte Reaktionen wie Kopfschmerzen, Schüttelfrost, hohen Blutdruck, Übelkeit, Asthenie und Schmerzen. Je 13 % der Patienten hatten Alopezie bzw. depressive Erkrankung, bei 8 % Fatigue. Bei je 5 % der Patienten traten unter Rituximab Pruritus, Urtikaria, schwerwiegende Depression, Reizbarkeit, Fieber, Oberbauchschmerzen, Tachykardie, muskuloskelettale Schmerzen und Papillome der Haut auf. Bei 37 % der Patienten unter Rituximab traten behandlungsbedingte Infektionen auf, im Vergleich zu 42 % der Patienten im Prednison-Arm. Die häufigsten Infektionen unter Rituximab waren Herpes-zoster- und -simplex-Infektionen, Bronchitis, Harnwegsinfektion, Pilzinfektion und Konjunktivitis.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, sollten der Handelsname und die Chargenbezeichnung des verabreichten Arzneimittels in der Patientenakte eindeutig dokumentiert werden und bei Meldungen von Nebenwirkungen angegeben werden.
Alle Patienten, die Rituximab erhalten, müssen einen Patientenpass erhalten, der bei jeder neuen Infusion kontrolliert bzw. ergänzt wird. Der Pass enthält für den Patienten wichtige Sicherheitsinformationen bezüglich eines möglicherweise erhöhten Infektionsrisikos einschließlich progressiver multifokaler Leukoenzephalopathie. Dieser ist zusammen mit dem behördlich beauflagten Schulungsmaterial, das dazu dient, die Wissensvermittlung zu optimieren und Hilfe bei der sicheren Anwendung des Arzneimittels zu geben, verfügbar unter: https://www.pei.de/DE/arzneimittelsicherheit-vigilanz/schulungsmaterial/schulungsmaterial-node.html.
Weiterführende Informationen
Rituximab (MabThera®) wurde für diese Indikation nicht in die Bewertung des Zusatznutzens nach § 35a SGB V vom G-BA aufgenommen.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) MabThera®, erschienen am 12. Juli 2019. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 2. September 2019 vorab online veröffentlicht.