Fremanezumab (Ajovy®) ▼
Zugelassene Indikation und Wirkmechanismus
Fremanezumab (Ajovy®) ist zur Migräneprophylaxe bei Erwachsenen mit mindestens vier Migränetagen pro Monat zugelassen.
Fremanezumab ist ein aus einer murinen Vorläuferzelle gewonnener humanisierter monoklonaler IgG2Δa/κ-Antikörper, der mittels rekombinanter DNA-Technik in Ovarialzellen des chinesischen Hamsters (Chinese Hamster Ovary, CHO) hergestellt wird. Er bindet selektiv das Calcitonin Gene-Related Peptide (CGRP) und hindert beide CGRP-Isoformen (α- und β-CGRP) an der Bindung an den CGRP-Rezeptor, der in der Pathophysiologie der Migräne eine zentrale Rolle spielt. CGRP ist ein Neuropeptid, das die nozizeptive Signalübertragung reguliert und als Vasodilatator wirkt. Der CGRP-Spiegel steigt während eines Migräneanfalls an und normalisiert sich beim Abklingen der Kopfschmerzen.
Fremanezumab wird subkutan am Abdomen, am Oberschenkel oder an der Außenseite des Oberarms oder in den Gesäßbereich appliziert. Patienten sollen nach angemessener Schulung Fremanezumab selbst verabreichen. Es stehen zwei Dosierungsoptionen zur Verfügung: 225 mg einmal monatlich (monatliche Dosierung) oder 675 mg alle drei Monate (vierteljährliche Dosierung). Bei einem Wechsel des Dosierungsplans sollte die erste Dosis des neuen Plans am nächsten geplanten Verabreichungstermin des vorherigen Dosierungsplans verabreicht werden.
Fremanezumab muss im Kühlschrank bei 2–8°C gelagert werden. Ungekühlt kann das Arzneimittel bis zu 24 Stunden bei bis zu 25°C gelagert werden.
Markteinführung
Fremanezumab (Ajovy®) ist seit dem 15. Mai 2019 in dieser Indikation auf dem deutschen Markt.
Bewertung
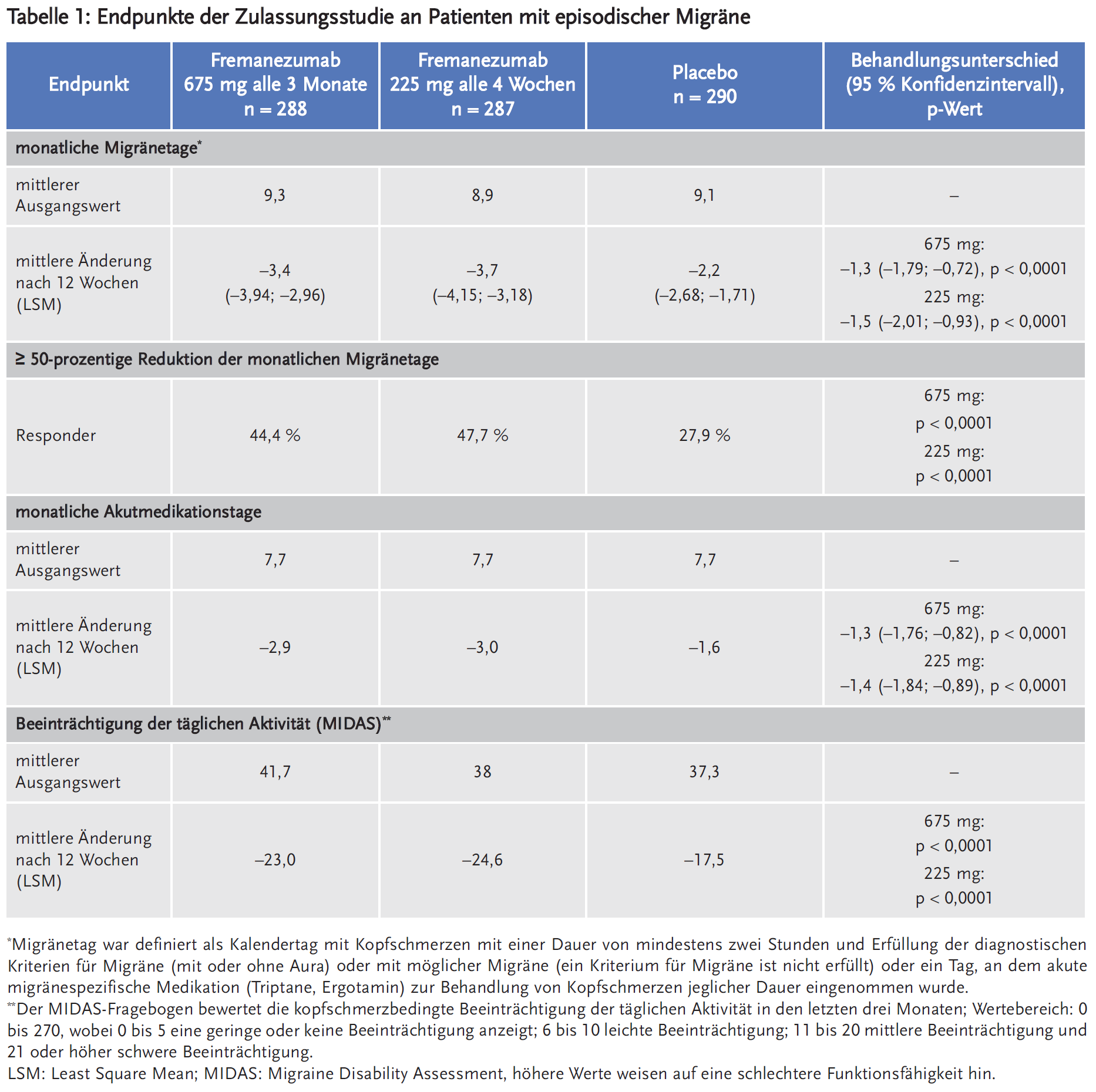
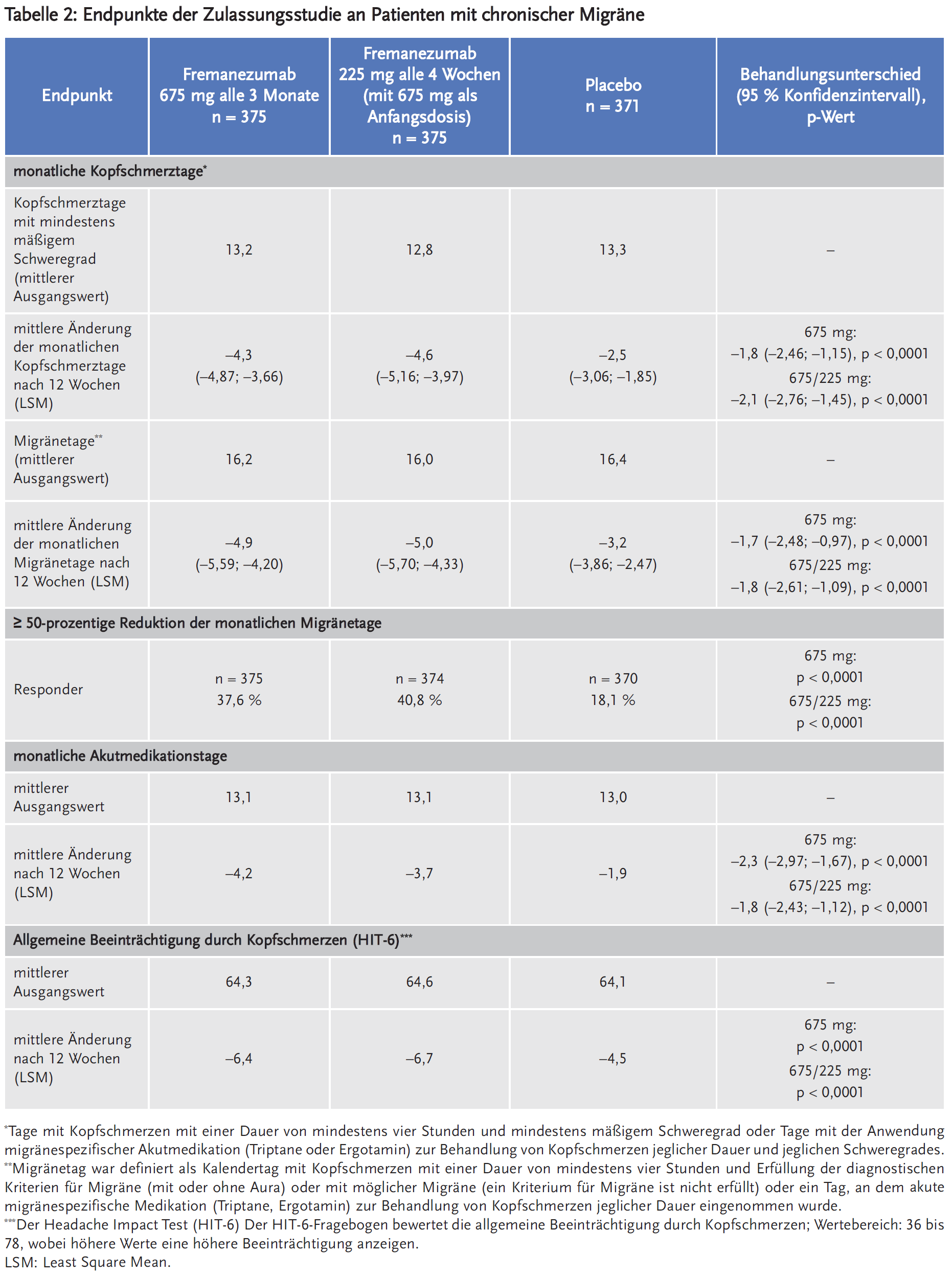
Fremanezumab ist der dritte monoklonale Antikörper, der sich spezifisch gegen das Migräne auslösende Neuropeptid Calcitonin-Gene-Related-Peptide (CGRP) richtet. Fremanezumab reduzierte in den Phase-III-Studien die durchschnittlichen Migränetage um 3,4–3,7 pro Monat (bei episodischer Migräne, EM) bzw. um 4,9–5,0 pro Monat (bei chronischer Migräne, CM). Placebo reduzierte die Migränetage um 2,2 bzw. 3,2 pro Monat. Der Unterschied zwischen Fremanezumab und Placebo erreichte statistische Signifikanz. Unter Fremanezumab wurde eine mindestens 50-prozentige Reduktion der monatlichen Migränetage bei 37,6–40,8 % der Patienten mit CM (versus 18,1 % unter Placebo; NNT 4–5 pro drei Monate) sowie bei 44,4–47,7 % der Patienten mit EM (versus 27,9 % unter Placebo; NNT = 5–6 pro drei Monate) berichtet. Fremanezumab verringerte zudem statistisch signifikant stärker als Placebo die Anzahl der Tage, an denen eine akute Migränemedikation erforderlich war: bei CM im Mittel um 3,7–4,2 Tage versus 1,9 Tage unter Placebo, bei EM im Mittel um 2,9–3 Tage versus 1,6 Tage unter Placebo. Nebenwirkungen wie lokale Reaktionen an der Injektionsstelle – Schmerzen, Verhärtung, Erythem – traten sehr häufig auf.
Fremanezumab bietet – wie bereits die ersten zwei CGRP-Rezeptorantagonisten Erenumab und Galcanezumab – gegenüber den verfügbaren Alternativen zur Migräneprophylaxe einen vergleichbaren Effekt. Der Vorteil gegenüber bisher verfügbaren Wirkstoffen scheint nach bisherigen Studiendaten in der besseren Verträglichkeit zu liegen. Ein weiterer Vorteil kann die monatliche und sogar vierteljährliche Applikation sein, die allerdings subkutan erfolgen muss. Es gibt keinen direkten Vergleich der drei verfügbaren Antikörper Erenumab, Galcanezumab und Fremanezumab. CGRP hat eine ausgeprägte vasodilatatorische Wirkung. Die Hemmung seines Rezeptors bzw. der Bindung der CGRP-Liganden birgt daher theoretisch das Risiko für kardiovaskuläre Ereignisse, das bei Migräne ohnehin gering erhöht ist. Die verfügbaren Studien ergaben keine eindeutigen Hinweise auf ein erhöhtes kardiovaskuläres Risiko, allerdings wurden Patienten > 70 Jahre oder mit kardiovaskulären Ereignissen in der Vorgeschichte ausgeschlossen. Die Risiken einer langfristigen Blockade von CGRP mit Fremanezumab – insbesondere hinsichtlich kardiovaskulärer Nebenwirkungen – können zum jetzigen Zeitpunkt nicht abschließend beurteilt werden, da Langzeitdaten zur Anwendung von Fremanezumab fehlen (die maximale Behandlungsdauer in den placebokontrollierten Zulassungsstudien war 181 Tage; in der nicht kontrollierten Langzeitstudie zwölf Monate). Der Einsatz von Fremanezumab sollte daher vorerst nur nach Versagen anderer Arzneimittel zur Migräneprophylaxe oder bei deren Unverträglichkeit erfolgen.
Wirksamkeit in den Zulassungsstudien
Fremanezumab wurde in zwei zwölfwöchigen, multizentrischen, doppelblinden, randomisierten, placebokontrollierten Phase-III-Studien an Patienten mit episodischer Migräne (EM) und mit chronischer Migräne (CM) untersucht.
Die Phase-III-Studie an Patienten mit EM (Studie 1) schloss 875 Patienten (742 Frauen, 133 Männer) ein, die in der Screeningphase 6–14 Kopfschmerztage pro Monat aufwiesen und an mindestens vier davon Migräne mit oder ohne Aura hatten oder die ein Triptan oder Ergotamin eingenommen haben. Ausgeschlossen wurden Patienten, die in den letzten vier Monaten vor Screening Botulinumtoxin angewendet haben, Opioide oder Barbiturate an mehr als vier Tage in der Screeningphase angewendet haben oder solche, die auf ≥ 2 medikamentöse Migräneprophylaktika nicht angesprochen haben. Die Patienten wurden in einen von drei Behandlungsarmen randomisiert: 675 mg Fremanezumab alle drei Monate und je eine Placebo-Injektion alle vier Wochen zwischen zwei Gaben von Fremanezumab (n = 291); Fremanezumab 225 mg einmal monatlich (n = 290) oder monatliche Verabreichung von Placebo (n = 294). Sie waren im Median 42 Jahre alt (Spanne: 18 bis 70 Jahre). Die mittlere Migränehäufigkeit bei Baseline betrug ca. neun Migränetage pro Monat. Die Anwendung akuter Kopfschmerzmedikation während der Studie war erlaubt, zudem führten etwa 21 % der Patienten ihre präventive Begleitmedikation (z. B. Betablocker, Kalziumantagonisten, Antidepressiva, Antikonvulsiva) fort.
Als primärer Endpunkt wurde die durchschnittliche Änderung der Anzahl der Migränetage pro Monat erhoben. Sekundäre Endpunkte waren u. a. die Ansprechrate (der Anteil der Patienten, die eine mindestens 50-prozentige Reduktion der Migränetage pro Monat erreichten) sowie die durchschnittliche Änderung der Anzahl der Tage mit akuter Kopfschmerzmedikation im Vergleich zum Studienbeginn. Zudem wurde die migränebedingte Beeinträchtigung der täglichen Aktivität anhand des Migraine-Disability-Assessment(MIDAS)-Fragebogens evaluiert.
Fremanezumab senkte in der Studie in beiden Dosierungen statistisch signifikant stärker als Placebo die Anzahl der monatlichen Migränetage sowie der Tage pro Monat mit Akutmedikation gegen Kopfschmerzen. Unter Fremanezumab erreichten statistisch signifikant mehr Patienten eine mindestens 50-prozentige Reduktion der monatlichen Migränetage als unter Placebo, allerdings waren das nur 44,4 bis 47,7 % der Patienten versus 27,9 % der Patienten unter Placebo. Dies entspricht einer Number needed to treat (NNT) von 5–6 pro drei Monate. Auch bezüglich der Beeinträchtigung der täglichen Aktivität war der Unterschied zu Placebo statistisch signifikant (Tabelle 1). Zwischen den zwei evaluierten Dosierungen zeigten sich keine relevanten Unterschiede in der Wirksamkeit.
In die HALO-Studie (Studie 2) wurden 1130 Patienten mit CM (991 Frauen, 139 Männer) in einen von drei Behandlungsarmen randomisiert: 675 mg Fremanezumab als Anfangsdosis, gefolgt von 225 mg Fremanezumab einmal monatlich (n = 379); 675 mg Fremanezumab alle drei Monate und je eine Placebo-Injektion alle vier Wochen zwischen zwei Gaben von Fremanezumab (n = 376) oder monatliche Injektion von Placebo (n = 375). Einschlusskriterien waren u.a. Migräne seit mindestens zwölf Monaten und ≥ 15 Kopfschmerztage/Monat und davon ≥ 8 Migränetage/Monat in den vorausgegangenen 28 Tagen. Maximal 30 % der Patienten mit einer prophylaktischen Migränemedikation für mindestens zwei Monaten vor Studieneinschluss durften diese fortführen. Ausgeschlossen wurden Patienten, die in den letzten vier Monaten vor Screening Botulinumtoxin angewendet haben, Opioide oder Barbiturate an mehr als vier Tagen in der Screeningphase angewendet haben oder solche, die auf zwei bis vier medikamentöse Migräneprophylaxen nicht angesprochen haben.
Die Patienten waren in den Fremanezumab-Armen im Median 40 Jahre alt und im Placebo-Arm im Median 41 Jahre alt. Die mittlere Migränehäufigkeit bei Baseline betrug ca. neun Migränetage pro Monat. Die Anwendung akuter Kopfschmerzmedikation während der Studie war erlaubt, zudem führten etwa 21 % der Patienten ihre präventive Begleitmedikation (z. B. Betablocker, Kalziumantagonisten, Antidepressiva, Antikonvulsiva) fort. Die mittlere Migränehäufigkeit bei Studienbeginn betrug etwa 16 Migränetage pro Monat, an im Mittel 11 Tagen im Monat erfolgte die Einnahme migränespezifischer Medikation.
Als primärer Endpunkt wurde die durchschnittliche Änderung der Anzahl der Kopfschmerztage pro Monat mit entweder mindestens mäßigem Schweregrad oder der Anwendung von migränespezifischer Akutmedikation (Triptane oder Ergotamin) erhoben. Sekundäre Endpunkte waren u. a. die Ansprechrate (der Anteil der Patienten, die eine mindestens 50-prozentige Reduktion der Migränetage pro Monat erreichten) sowie die durchschnittliche Änderung der Migränetage pro Monat und der Anzahl der Tage mit akuter Kopfschmerzmedikation im Vergleich zum Studienbeginn. Zudem wurde die allgemeine Beeinträchtigung durch Kopfschmerz anhand des Six-item-Headache-Impact-Test(HIT-6)-Fragebogens evaluiert.
Auch in dieser Studie zeigten sich signifikante Unterschiede unter beiden Dosierungsregimen von Fremanezumab im Vergleich zu Placebo bezüglich patientenrelevanter Wirksamkeitsendpunkte wie u. a. der Reduktion der monatlichen Akutmedikationstage, der Ansprechrate sowie der Beeinträchtigung der täglichen Aktivität (Tabelle 2). Fremanezumab reduzierte die Anzahl der durchschnittlichen monatlichen Migränetage um 4,9–5 Tage sowie der Kopfschmerztage mit mindestens mäßigem Schweregrad um 4,3–4,6 Tage. Der Unterschied zu Placebo war statistisch signifikant. Unter Fremanezumab erreichten allerdings nur 37,6 bzw. 40,8 % der Patienten eine mindestens 50-prozentige Reduktion der monatlichen Migränetage versus 18,1 % der Patienten unter Placebo. Dies entspricht einer NNT von 4–5 pro drei Monate. Auch bezüglich der allgemeinen Beeinträchtigung durch Kopfschmerzen (HIT-6) war der Unterschied zu Placebo statistisch signifikant. Zwischen den zwei evaluierten Dosierungsregime zeigten sich keine relevanten Unterschiede in der Wirksamkeit.
Die Patienten, die die Phase-III-Zulassungsstudien abgeschlossen hatten, wurden zusammen mit weiteren 300 Patienten (jeweils etwa die Hälfte davon mit CM bzw. EM) in eine doppelblinde Langzeitstudie eingeschlossen. Sie (n = 1889) wurden im Verhältnis 1:1 auf folgende zwei Behandlungsarme randomisiert: Patienten mit CM bekamen eine Initialgabe von 675 mg Fremanezumab und elf monatliche 225-mg-Fremanezumab-Dosen oder 675 mg vierteljährlich für zwölf Monate, während Patienten mit EM 225 mg Fremanezumab monatlich oder 675 mg vierteljährlich erhielten. Evaluiert wurden die Langzeitsicherheit und Verträglichkeit von Fremanezumab. Insgesamt sollen 79 % der Patienten den zwölfmonatigen Behandlungszeitraum der Studie abgeschlossen haben. Über die zwei Dosierungsschemata gepoolt soll nach 15 Monaten eine Reduktion der monatlichen Migränetage von 6,6 gegenüber Baseline in den pivotalen Studie 1 und Studie 2 erreicht worden sein. Die 50-prozentige Ansprechrate am Studienende soll 61 % betragen haben. Zudem sollen sich im Behandlungszeitraum keine Sicherheitssignale ergeben haben1.
Ausgewählte Nebenwirkungen
Sehr häufig treten lokale Reaktionen an der Injektionsstelle auf: Schmerzen, Verhärtung, Erythem; Juckreiz an der Injektionsstelle häufig; Ausschläge an der Injektionsstelle gelegentlich.
In den Zulassungsstudien wurden drei Todesfälle berichtet, einer war zwar zerebrovaskulärer Genese, trat aber erst 300 Tage nach der letzten verabreichten Fremanezumab-Dosis auf. Schwerwiegende kardiovaskuläre unerwünschte Ereignisse traten in den placebokontrollierten Phase-III-Studien unter Fremanezumab und Placebo gleich häufig bei insgesamt weniger als 1 % der Patienten auf. Hypertension wurde bei elf Patienten unter Fremanezumab und vier Patienten unter Placebo berichtet, Tachykardie bei jeweils drei Patienten, Palpitationen und erhöhte Herzrate bei je drei Patienten unter Fremanezumab und zwei Patienten unter Placebo und hypertensive Krisen bei zwei Patienten unter Fremanezumab und keinem Patienten unter Placebo. In der Sicherheitskohorte hatten 56 % mindestens einen kardiovaskulären oder zerebrovaskulären Risikofaktor, am häufigsten waren darunter Adipositas, Anwendung hormoneller Kontrazeptiva, kardiovaskuläre Erkrankung in der Vergangenheit, Bluthochdruck und Fettstoffwechselstörungen.
Bei den Patienten im Alter von 65 bis 70 Jahren traten unter Fremanezumab unerwünschte Ereignisse unabhängig von dem Organsystem numerisch häufiger auf als bei jüngeren Patienten. Während der doppelblinden Langzeitstudie kam es bei zwei Patienten zu tiefen Venenthrombosen sowie bei jeweils einem Patienten zu einer transitorischen ischämischen Attacke, Hypertension und Venenthrombose der Extremitäten.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
Ältere Patienten (> 75 Jahre) wurden in die Studie nicht eingeschlossen, im Alter von 65 bis 70 Jahren wurden lediglich 61 von insgesamt 2512 Patienten in allen placebokontrollierten Studien (Phase II und III) sowie in der Langzeitstudie mit Fremanezumab behandelt.
Von der Teilnahme an den klinischen Studien waren u. a. ausgeschlossen: Patienten mit vorbestehendem Myokardinfarkt, Schlaganfall, tiefen Venenthrombosen, transitorischen ischämischen Attacken und Lungenembolie sowie Patienten mit einer schwerwiegenden kardiovaskulären Erkrankung. Für diese Patientengruppen liegen weder Wirksamkeits- noch Sicherheitsdaten vor.

Weiterführende Informationen
Das IQWiG wurde am 15.05.2019 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Ajovy®, erschienen am 17. April 2019. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Fußnote
1Die Studie ist nicht veröffentlicht, zum Zeitpunkt der Zulassung lief sie noch, sodass für die Zulassung der Datenschnitt vom 31.05.2017 berücksichtigt wurde. Die Angaben stammen aus der deutschsprachigen Produktinformation (Datum Veröffentlichung 17.04.2019).
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 9. Juli 2019 vorab online veröffentlicht.