Tenofoviralafenamid (Vemlidy®) (frühe Nutzenbewertung)
Update – Neue Arzneimittel
Update – Neue Arzneimittel
Update – Neue Arzneimittel
Die Hepatitis B ist eine akut oder chronisch verlaufende Infektionskrankheit der Leber, die durch Hepatitis-B-Viren (HBV) ausgelöst wird. Die Übertragung erfolgt überwiegend sexuell, durch Kontakt mit kontaminiertem Blut oder Körperflüssigkeiten, durch unsterile Instrumente (u. a. sog. „needle sharing“ beim i.v.-Drogenkonsum) und perinatal. Eine akute HBV-Infektion verläuft bei 80–90 % der Erwachsenen selbstlimitierend unter Bildung von Antikörpern; etwa 70 % dieser selbstlimitierenden Infektionen verlaufen klinisch unbemerkt. Bei etwa 0,5–1 % der nicht geimpften Erwachsenen ist der Verlauf fulminant bis tödlich, in ca. 5–10 % der Fälle chronifiziert die Infektion. Von diesen entwickelt etwa die Hälfte eine chronische Entzündung, deren mögliche Folgen Leberzirrhose und hepatozelluläres Karzinom sind (1).
Tenofoviralafenamid (Vemlidy®) ist ein Phosphonamidat-Prodrug von Tenofovir (2’-Desoxyadenosinmonophosphat-Analogon). Der Substanzeintritt in die primären Hepatozyten erfolgt durch passive Diffusion sowie über die hepatischen Aufnahmetransporter OATP1B1 und OATP1B3. In den Hepatozyten wird sie durch das Enzym Carboxylesterase 1, in den mononukleären Zellen des peripheren Blutes durch die Serinprotease Cathepsin A zunächst zu Tenofovir hydrolysiert. Anschließend wird Tenofovir zum pharmakologisch aktiven Metaboliten Tenofovirdiphosphat phosphoryliert. Tenofovirdiphosphat wird durch die viruseigene reverse Transkriptase in die virale DNA eingebaut und führt einen DNA-Kettenabbruch herbei. Dadurch wird die Virusreplikation gehemmt (2). Tenofovir wirkt spezifisch auf das HBV und das humane Immundefizienzvirus (HIV-1 und HIV-2) und wird auch in Kombination mit anderen antiretroviralen Arzneimitteln zur Behandlung von Erwachsenen und Jugendlichen mit HIV-1-Infektion eingesetzt.
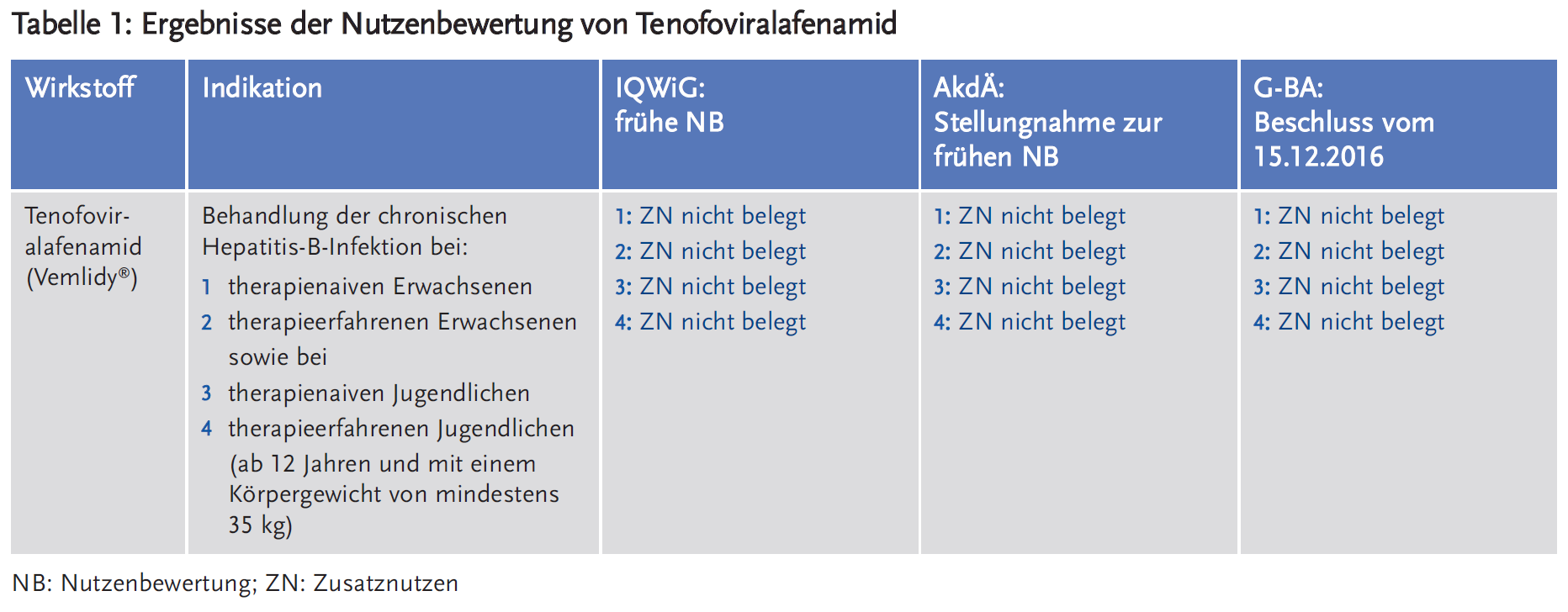
Aus der Festlegung der ZVT des G-BA ergaben sich vier Fragestellungen (d. h. therapeutische Situationen) zur Bewertung des Zusatznutzens von TAF in folgenden Patientengruppen:
Das IQWiG bemängelte die vom pU vorgenommene Zuordnung von therapienaiven und vorbehandelten Patienten jeweils in „oral antiviral unvorbehandelte“ und „oral antiviral vorbehandelte“ Patienten. Parenteral mit Interferon vorbehandelte Patienten wurden einer anderen Fragestellung zugeordnet als vom G-BA festgelegt. Die vorgelegten Daten aus zwei randomisierten, doppelblinden Studien waren aus Sicht des IQWiG für die Ableitung des Zusatznutzens nicht geeignet, weil sie inhaltlich unvollständig waren und durch weitere Limitationen eine eingeschränkte Interpretierbarkeit aufwiesen (3).
Die AkdÄ stimmte der Kritik des IQWiG bezüglich der Fokussierung auf Schadwirkungen von TAF auf Nierenfunktion (Erkrankungen der Nieren und Harnwege) und Knochenstoffwechsel (Veränderungen der Knochendichte/Frakturen) durch den pU zu. Die hierbei gewählten Surrogatvariablen sind als Endpunkte für die Vorhersage eines langfristigen klinischen Effektes nicht geeignet. Die Verschlechterung der Kreatinin-Clearance mit der verwendeten Methode (geschätzte glomeruläre Filtrationsrate nach Cockcroft-Gault) ließ aus Sicht der AkdÄ nur eine Beurteilung der aktuellen Nierenfunktion, nicht aber eine Aussage über die Entwicklung einer zukünftigen dauerhaften Beeinträchtigung der Nierenfunktion zu. Die beobachtete geringe Abnahme der Knochendichte ist ebenfalls ungeeignet, die zu erwartende Frakturrate als klinisch relevanten Endpunkt vorherzusagen. Auch wenn die vorgelegten Studien eine geringere Beeinträchtigung der Nierenfunktion und des Knochenstoffwechsels durch TAF im Vergleich zu TDF andeuten, sind sie nicht geeignet, eine verlässliche Langzeitprognose bezüglich klinisch relevanter Nierenfunktionsstörungen oder Knochenbrüchen zu abzuleiten. Des Weiteren wies die AkdÄ auf mögliche Unterschiede bezüglich neurologischer unerwünschter Wirkungen zuungunsten von TAF sowie auf die Veränderungen des Fettstoffwechsels unter TAF hin, die bezüglich kardiovaskulärer Folgewirkung bei einer über Jahrzehnte durchzuführenden Behandlung durchaus Relevanz erlangen können, insbesondere auch weil es aktuelle Hinweise auf einen möglicherweise kardioprotektiven Effekt von TDF gibt (4).
Der G-BA folgte der Dossierbewertung vom IQWiG und der Stellungnahme der AkdÄ und beschloss, dass ein Zusatznutzen von TAF nicht belegt ist. Die Entscheidung wurde mit der mangelnden, bruchstückhaften Ergebnispräsentation durch den pU bezüglich der selektiven Darstellung spezifischer oder häufiger unerwünschter Ereignisse begründet. Damit war es dem G-BA nicht möglich, alle für die Bewertung des Zusatznutzens relevanten Daten lückenlos und inhaltlich im Dossier des pU nachzuvollziehen und eine Beurteilung des Zusatznutzens bzw. eines möglichen Schadens abzugeben. Für die Patientengruppe der Jugendlichen wurden keine Ergebnisse aus klinischen Studien für die Nutzenbewertung vorgelegt (5).
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
Dieser Artikel wurde am 20. November 2017 vorab online veröffentlicht.