Nirmatrelvir/Ritonavir (Paxlovid®) ▼
Zugelassene Indikation und Wirkmechanismus
Paxlovid® (Nirmatrelvir/Ritonavir) ist zugelassen zur Behandlung der Coronavirus-Krankheit 2019 (COVID-19) bei Erwachsenen, die keine zusätzliche Sauerstoffzufuhr benötigen und ein erhöhtes Risiko haben, einen schweren COVID-19-Verlauf zu entwickeln.
Nirmatrelvir ist ein Inhibitor der 3-CL(„chymotrypsin-like cysteine“)-Protease und stört hierdurch die Replikation von SARS-CoV-2. Der vor allem aus der Therapie von HIV (Humanes Immundefizienz-Virus) bekannte Wirkstoff Ritonavir dient lediglich der Hemmung des CYP3A-vermittelten Abbaus von Nirmatrelvir. Durch die höheren Plasmaspiegel soll die Wirksamkeit von Nirmatrelvir verbessert werden.
Markteinführung
Paxlovid® (Nirmatrelvir/Ritonavir) erhielt am 28. Januar 2022 eine bedingte Zulassung. Der Bund hat eine Million Therapiezyklen beim Anbieter reserviert und sieht ein spezielles Verfahren zur Verordnung und Belieferung vor. Bei positivem Testergebnis können Ärzte eine Verordnung für Nirmatrelvir/Ritonavir direkt an die Apotheke übermitteln. Die Apotheke bestellt anschließend Nirmatrelvir/Ritonavir beim Großhandel und gibt es schnellstmöglich zusammen mit der Gebrauchsinformation an den Patienten ab (1).
Bewertung
Die Zulassungsstudie EPIC-HR („Evaluation of Protease Inhibition for Covid-19 in High-Risk Patients“) (2) untersuchte Nirmatrelvir/Ritonavir bei ambulanten Patienten mit COVID-19, die ein erhöhtes Risiko für einen schweren Verlauf hatten. Die Behandlung wurde innerhalb von fünf Tagen nach dem Auftreten von Symptomen begonnen. Nirmatrelvir/Ritonavir beeinflusste signifikant den kombinierten Endpunkt aus Gesamtmortalität und COVID-19-bedingten Hospitalisierungen (1 % vs. 6 %, Number needed to treat (NNT) = 18).
Patienten waren von der Studie ausgeschlossen, wenn sie gegen SARS-CoV-2 geimpft waren oder eine frühere SARS-CoV-2 Infektion bekannt war. Dennoch hatte die Hälfte der Patienten bei Studienbeginn einen positiven Antikörpernachweis. Schwere Verläufe waren bei diesen Patienten selten und unterschieden sich nur geringfügig zwischen den Armen (0,2 % vs. 1,5 %; NNT = 77). Bei geimpften oder genesenen Patienten ist wegen des niedrigeren Ausgangsrisikos von einem deutlich geringeren Nutzen von Nirmatrelvir/Ritonavir auszugehen.
Die Studie EPIC-HR wurde bei Patienten durchgeführt, die mit der Delta-Variante infiziert waren. Auch wenn Mutationen überwiegend das Spike-Protein und nicht die Virusproteasen betreffen, ist die Wirksamkeit von Nirmatrelvir/Ritonavir gegenüber der aktuell dominierenden Omikron-Variante unklar. Zudem führt die Omikron-Variante zu leichteren Krankheitsverläufen als die Delta-Variante (3). Durch das niedrigere Ausgangsrisiko ist bei Omikron gegenwärtig eine geringere absolute Risikoreduktion (ARR) durch Nirmatrelvir/Ritonavir zu erwarten.
In Subgruppenanalysen profitierten ältere Patienten (≥ 65 Jahre) am stärksten von Nirmatrelvir/Ritonavir (ARR 14 %; NNT 8). Allerdings sind bei diesen häufig multimorbiden Patienten zahlreiche Wechselwirkungen von Ritonavir mit der Begleitmedikation zu beachten (4;5). Zudem liegen keine klinischen Daten für Patienten mit moderater Niereninsuffizienz vor. Das Monitoring ist in der Praxis dadurch erschwert, dass die Patienten ihre Therapie mit Nirmatrelvir/Ritonavir in häuslicher Quarantäne erhalten.
Insgesamt ist in der aktuellen Versorgungssituation aufgrund des Vorherrschens von Omikron-Varianten und des relevanten Anteils (durch asymptomatische Infektionen) Immunisierter von einer geringeren absoluten Risikoreduktion für den untersuchten Endpunkt auszugehen als in der Studie EPIC-HR. Aus Sicht der AkdÄ erweitert Nirmatrelvir/Ritonavir die Therapieoptionen bei COVID-19 insbesondere für ältere Patienten ohne bekannte Immunität durch Impfung oder Genesung, da in dieser Patientengruppe weiterhin von einem erhöhten Ausgangsrisiko auszugehen ist (siehe auch (6)). Die Wirksamkeit von Nirmatrelvir/Ritonavir bei immunsupprimierten Patienten oder in Kombination mit monoklonalen Antikörpern lässt sich anhand der vorliegenden Daten nicht beurteilen.
Die Erfahrungen mit Remdesivir, aber auch mit Neuraminidasehemmern bei Influenza, sprechen für eine schwächere Wirksamkeit antiviraler Arzneimittel bei spätem Behandlungsbeginn (z. B. später als fünf Tage nach Infektion (7)). Für Nirmatrelvir/Ritonavir liegen ausschließlich Daten zu einem Behandlungsbeginn innerhalb von fünf Tagen nach dem Auftreten erster Symptome vor. Innerhalb dieses Zeitfensters nimmt die Wirksamkeit von Nirmatrelvir/Ritonavir geringfügig ab. Aus Sicht der AkdÄ sollten geeignete Hochrisikopatienten vorausschauend über die Therapieoption mit Nirmatrelvir/Ritonavir aufgeklärt werden, damit diese bereits bei ersten Symptomen einen Schnelltest auf SARS-CoV-2 durchführen und ihren Arzt kontaktieren (8).
Wirksamkeit in den Zulassungsstudien
Die Zulassung von Nirmatrelvir/Ritonavir basiert auf der doppelblinden, randomisierten, placebokontrollierten Phase-II/III-Studie EPIC-HR. Die Studienteilnehmer erhielten über fünf Tage alle zwölf Stunden jeweils 300 mg Nirmatrelvir und 100 mg Ritonavir (1120 Patienten) oder entsprechende Placebopräparate (1126 Patienten). Eingeschlossen wurden ambulante Patienten mit nachgewiesener SARS-CoV-2-Infektion, deren Symptombeginn maximal fünf Tage zurück lag und die mindestens einen Risikofaktor für einen schweren Krankheitsverlauf aufwiesen. Als Risikofaktoren galten unter anderem ein Alter ≥ 60 Jahre, Übergewicht (BMI > 25 kg/m²), Nikotinkonsum, Diabetes mellitus, Hypertonus, Immunsuppression, onkologische Erkrankungen sowie chronische renale, pulmonale oder kardiovaskuläre Erkrankungen. Männer und Frauen wurden zu gleichen Teilen eingeschlossen. Die Patienten waren median 46 Jahre alt. 30 % der Patienten kamen aus Europa und 41 % aus den USA. Die häufigsten Risikofaktoren waren Übergewicht (80 %), Nikotinkonsum (39 %), Hypertonus (33 %) und Diabetes mellitus (12 %). Zum Zeitpunkt der Studiendurchführung (Juli bis Dezember 2021) war die Delta Variante vorherrschend (Nachweis der Delta-Variante bei 99 % der Patienten).
Eine Studienteilnahme war nicht möglich, wenn die Patienten gegen SARS-CoV-2 geimpft oder bereits zu einem früheren Zeitpunkt mit SARS-CoV-2 infiziert waren. Trotz dieser Ausschlusskriterien wiesen die Hälfte der Patienten zum Zeitpunkt der Randomisierung einen positiven Antikörpertiter auf. Eine Differenzierung zwischen IgG und IgM Antikörpern erfolgte nicht. Es ist unklar, ob der Antikörpernachweis auf einer früheren, nicht diagnostizierten SARS-CoV-2-Infektion beruhte oder Ausdruck einer aktuellen Immunreaktion gegen SARS-CoV-2 war. Laut aktuellem Forschungsstand findet eine Serokonversion im Mittel in der zweiten Woche nach Symptombeginn statt (Range 4–22 Tage) (9). Es ist deshalb anzunehmen, dass ein relevanter Anteil der Patienten vor Studieneinschluss unbemerkt von COVID-19 genesen war.
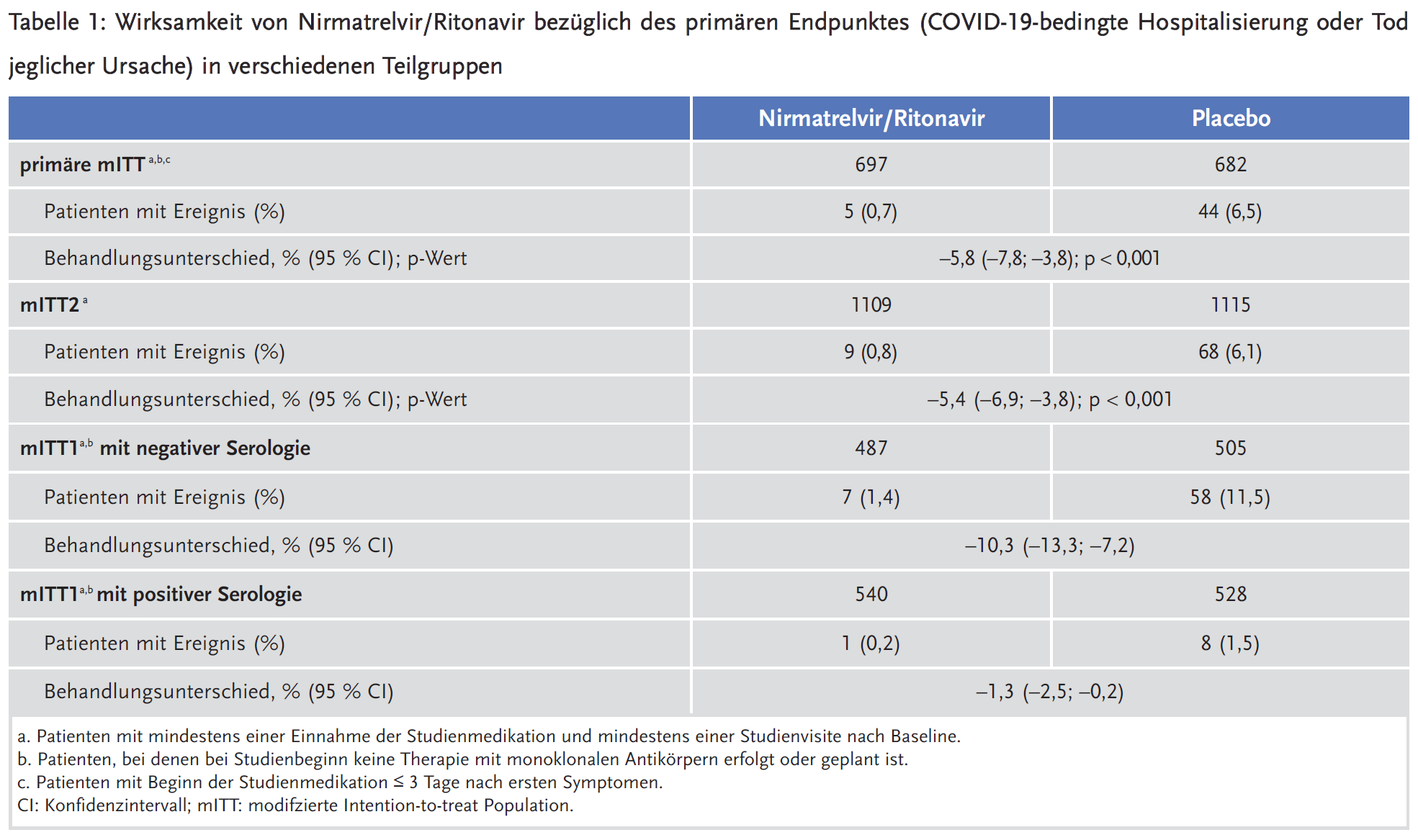
Primärer Endpunkt der Studie war der Anteil der Patienten, die innerhalb von 28 Tagen aufgrund von COVID-19 stationär behandelt wurden oder – gleich welcher Ursache – verstarben. Insgesamt ereigneten sich 12 Todesfälle, keiner davon unter Nirmatrelvir/Ritonavir. Der kombinierte Endpunkt wurde überwiegend durch COVID-19-bedingte Hospitalisierungen beeinflusst. In den vorliegenden Unterlagen werden keine Kriterien angegeben, die – in Abgrenzung zu einer Hospitalisierung mit COVID-19 – für eine COVID-19-bedingte Hospitalisierung erfüllt sein müssen. Ergänzende Angaben zur Hospitalisierungsrate jeglicher Ursache wären wünschenswert gewesen.
Die mITT(„modified intention-to-treat“)-Population umfasste Patienten mit mindestens einer Einnahme der Studienmedikation und mindestens einer Studienvisite nach Baseline. Je nach Beginn der Studienmedikation (≤ 3 oder ≤ 5 Tage nach ersten Symptomen) und dem Erhalt von monoklonalen Antikörpern wurden verschiedene Teilgruppen der mITT-Population unterschieden. Primär wurden Patienten der mITT-Population untersucht, deren Behandlung innerhalb von drei Tagen begann und bei denen zu Studienanfang keine Therapie mit monoklonalen Antikörpern erfolgt bzw. geplant war.
Aufgrund des positiven Ergebnisses einer geplanten Zwischenauswertung wurde die Rekrutierung nach Einschluss von 2246 Patienten abgebrochen (entsprechend 75 % der ursprünglich geplanten Patientenzahl). In der finalen Auswertung erfüllten 1379 Patienten die Kriterien der primären ITT-Population. Bei diesen Patienten führte Nirmatrelvir/Ritonavir zu einer absoluten Risikoreduktion für COVID-19-bedingte Hospitalisierungen oder Tod (NNT = 18) von knapp 6 %. Eine ähnliche Effektivität bestand auch in der mITT2-Population (n = 2224). Diese umfasste sowohl Patienten mit einem späteren Behandlungsbeginn (Tag 4 oder 5 nach Symptombeginn) als auch Patienten mit zusätzlicher Antikörpertherapie.
Subgruppenanalysen stützen die Annahme, dass der Behandlungsbeginn nicht wesentlich die Wirksamkeit von Nirmatrelvir/Ritonavir beeinflusst (≤ 3 Tage vs. > 3 Tage: ARR 5,8 % vs. 5,2 %). Dagegen lässt sich die Wirksamkeit von Nirmatrelvir/Ritonavir bei zusätzlicher Gabe von monoklonalen Antikörpern nicht sicher beurteilen, da die Anzahl der Patienten gering (n = 139) und ihre Ereignisrate niedrig war (ein Ereignis unter Nirmatrelvir/Ritonavir, zwei Ereignisse unter Placebo). Weitere Subgruppenanalysen zeigten eine weitgehend konstante relative Risikoreduktion (84–95 %) durch Nirmatrelvir/Ritonavir bei Vorliegen typischer Risikofaktoren (höheres Alter, Adipositas) bzw. protektiver Faktoren (jüngeres Alter, positive Serologie). Entsprechend dem unterschiedlichen Ausgangsrisiko schwankte die Höhe der absoluten Risikoreduktion erheblich: Während die absolute Risikoreduktion bei älteren Patienten (≥ 65 Jahre) 14 % betrug (NNT = 8), lag sie bei Patienten mit positiver Serologie nur wenig über 1 % (NNT = 77).
Ausgewählte Nebenwirkungen
Die häufigsten unerwünschten Ereignisse (UE) unter Nirmatrelvir/Ritonavir waren Störungen des Geruchs- und Geschmackssinnes (6 %) sowie Durchfall (3 %). Hypertonus und Muskelschmerzen traten selten (< 1 %), aber numerisch häufiger unter Nirmatrelvir/Ritonavir auf. Alle genannten UE sind bekannte Risiken von Ritonavir.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Durch Hemmung von CYP3A beeinflusst Ritonavir die Plasmaspiegel zahlreicher Arzneimittel. Relevante Wechselwirkungen können bis zu fünf Tage nach Einnahme von Ritonavir auftreten. Vor einer Verordnung von Nirmatrelvir/Ritonavir muss die aktuelle Komedikation erfragt und auf Wechselwirkungen überprüft werden. Es sind zahlreiche, breit eingesetzte Substanzgruppen betroffen. Aufgrund ausgeprägter Interaktionen kann Nirmatrelvir/Ritonavir kontraindiziert sein (4,5).
- Die Studie EPIC-HR schloss sehr wenige (< 1 %) Patienten mit Immundefizienz ein. Es ist aktuell unklar, ob die empfohlene Behandlungsdauer von fünf Tagen bei immungeschwächten Patienten das Risiko für Therapieversagen erhöht und die Entwicklung von Resistenzen fördert.
- Patienten mit mäßiger bis schwerer Niereninsuffizienz waren aus der Studie EPIC-HR ausgeschlossen. Eine pharmakokinetische Studie zeigte einen Anstieg von UE bei zunehmender Niereninsuffizienz. Der pharmazeutische Unternehmer empfiehlt bei moderat eingeschränkter Nierenfunktion eine Halbierung der Dosis (150 mg/100 mg). Für diese Dosisanpassung liegen jedoch keine Daten aus klinischen Studien vor. Die Gabe von Nirmatrelvir/Ritonavir bei schwerer Niereninsuffizienz wird nicht empfohlen.
- Für Patienten mit schwerer Leberinsuffizienz liegen keine Daten zu Nutzen und Risiken von Nirmatrelvir/Ritonavir vor. Die Zwischenauswertung einer pharmakokinetischen Studie zeigte keine abweichenden Ergebnisse für Patienten mit moderater Leberinsuffizienz im Vergleich zu Patienten mit normaler Leberfunktion.

Weiterführende Informationen
Nirmatrelvir/Ritonavir (Paxlovid®) wurde bislang noch nicht in die Bewertung des Zusatznutzens nach § 35a SGB V vom G-BA aufgenommen.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Paxlovid®, erschienen am 24. Februar 2022. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Literatur
- Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM): Informationen zu Lagevrio® und Paxlovid®: www.bfarm.de/DE/Arzneimittel/Arzneimittelinformationen/covid-19-arzneimittel.htm. Letzter Zugriff: 23. März 2022.
- Hammond J, Leister-Tebbe H; Garnder A et al., EPIC-HR Investigators: Oral nirmatrelvir for high-risk, nonhospitalized adults with covid-19. N Engl J Med 2022; 386: 1397-1408.
- Kahn F, Bonander C, Moghaddassi M et al.: Risk of severe COVID-19 from the Delta and Omicron variants in relation to vaccination status, sex, age and comorbidities – surveillance results from southern Sweden, July 2021 to January 2022. Euro Surveill 2022; 27: 2200121.
- Fachgruppe COVRIIN am Robert Koch-Institut: Hinweise zu Arzneimittelwechselwirkungen von Paxlovid® (Nirmatrelvir/Ritonavir): www.rki.de/DE/Content/InfAZ/N/Neuartiges_Coronavirus/COVRIIN_Dok/Arzneimittelwechselwirkungen_Paxlovid.pdf (letzter Zugriff: 23. März 2022). Erscheinungsdatum: 10. Februar 2022.
- European Medicines Agency (EMA): European Public Assessment Report (EAPR) Paxlovid® – Summary of product characteristics: www.ema.europa.eu/en/documents/product-information/paxlovid-epar-product-information_en.pdf (letzter Zugriff: 23. März 2022). Stand: 28. Januar 2022.
- Mühlbauer B, Schott G, Ludwig W-D: Zum klinischen Nutzen von Molnupiravir und Nirmatrelvir in der Behandlung nicht hospitalisierter Patienten mit COVID-19 und einem Risiko für einen schweren Verlauf. Arzneiverordnung in der Praxis (AVP)2022: vorab online, 11. März 2022.
- Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ): Neue Arzneimittel: Remdesivir (Veklury®). Arzneiverordnung in der Praxis (AVP) 2021; 48: 108-113.
- Salisbury H: Are antivirals a covid-19 game changer? BMJ 2022; 376: o810.
- Arbeitsgemeinschaft zur Coronavirus-Diagnostik, Institut für Virologie der Charité und Robert Koch- Institut: Antikörpernachweise zum Nachweis stattgehabter Infektionen: www.rki.de/DE/Content/InfAZ/N/Neuartiges_Coronavirus/Vorl_Testung_nCoV.html;jsessionid=968A9476AC95DCDD266188370A8FC1CC.internet092 Letzter Zugriff: 23. März 2022.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 26. April 2022 vorab online veröffentlicht.