Vericiguat (Verquvo®) ▼
Zugelassene Indikation
Verquvo® (Vericiguat) ist zugelassen zur Behandlung von symptomatischer, chronischer Herzinsuffizienz bei erwachsenen Patienten mit reduzierter Ejektionsfraktion, die nach einem kürzlich aufgetretenen Dekompensationsereignis, das eine i.v.-Therapie erforderte, stabilisiert wurden.
Herzinsuffizienz und pulmonal-arterielle Hypertonie (PAH) sind mit einer verminderten Synthese von vasodilatierend wirkendem Stickstoffmonoxid (NO) assoziiert. Vericiguat stimuliert die lösliche Guanylatzyklase (sGC) und steigert hierdurch die Synthese von cyclischem Guanosinmonophosphat (cGMP). Ein erhöhter intrazellulärer cGMP-Spiegel führt unabhängig von NO zu einer Vasodilatation der glatten Muskulatur. Vericiguat soll auch die Bindungsstelle von sGC für endogenes NO sensibilisieren, dem physiologischen Aktivator von sGC. Darüber hinaus werden antiproliferative, antiinflammatorische und antifibrotische Effekte von cGMP diskutiert.
Vericiguat ist der zweite zugelassene sGC-Stimulator. Riociguat wurde – mit gleichem Wirkmechanismus – 2014 zugelassen zur Behandlung der PAH.
Markteinführung
Verquvo® (Vericiguat) ist seit 15.09.2021 in dieser Indikation auf dem deutschen Markt verfügbar.
Bewertung
Die Zulassungsstudie VICTORIA untersuchte Vericiguat bei Patienten mit HFrEF (Heart Failure with reduced Ejection Fraction), deren letzte Dekompensation durchschnittlich etwa einen Monat zurück lag und überwiegend stationär behandelt worden war. Primärer Endpunkt der Studie war die Kombination aus kardiovaskulärem Tod oder Hospitalisierung aufgrund von Herzinsuffizienz. Vericiguat führte zu einer statistisch signifikanten, aber klinisch allenfalls moderaten Risikoreduktion im Vergleich zu Placebo (35,5 % vs. 38,5 %), die vor allem auf einer Verringerung der herzinsuffizienzbedingten Hospitalisierungen beruhte. Die Gesamtmortalität wurde nicht signifikant beeinflusst. Unter Vericiguat traten häufiger symptomatische Hypotonien und Anämien auf als unter Placebo.
Vericiguat sollte laut Studienprotokoll zusätzlich zu einer Standardtherapie der Herzinsuffizienz gegeben werden. Trotz schwerer Erkrankung erhielten allerdings nur 60 % der Patienten eine voll-ständige Basistherapie, bestehend aus ACE(Angiotensin Converting Enzyme)-Hemmer bzw. ARB (Angiotensinrezeptorblocker), Betarezeptorenblocker und MRA (Mineralokortikoidrezeptor-Antagonisten). Eine Intensivierung der Medikation mit ARNI (Angiotensin-Rezeptor-Neprilysin-Inhibitoren) oder SGLT-2(Natrium-Glukose-Cotransporter-2)-Inhibitoren erfolgte nur bei wenigen Patienten. Angaben zur Diuretikatherapie zu Studienbeginn und im Verlauf liegen nicht vor. Die Effektivität von Vericiguat als Add-on zu einer optimierten, leitliniengerechten Herzinsuffizienztherapie lässt sich deshalb aus den vorliegenden Daten nicht abschließend beurteilen.
Subgruppenanalysen zeigten für Patienten mit sehr hohen NT-proBNP(N-terminal pro-Brain Natriuretic Peptide)-Ausgangswerten unter Vericiguat sogar ein numerisch höheres Risiko für kardiovaskulären Tod oder Hospitalisierung aufgrund von Herzinsuffizienz. Daher schränkt die europäische Arzneimittel-Agentur (EMA) die Zulassung von Vericiguat auf Patienten ein, die nach ihrer Dekompensation stabilisiert wurden.
Aus Sicht der AkdÄ kann aktuell keine abschließende Empfehlung für Vericiguat bei Patienten mit HFrEF und stattgehabter Dekompensation ausgesprochen werden. Im Einzelfall könnte Vericiguat eine Option bei diesen schwer erkrankten Patienten darstellen, wenn eine bestmögliche Umsetzung der Herzinsuffizienztherapie entsprechend den aktuellen Leitlinien erfolglos bleibt.
Wirksamkeit in den Zulassungsstudien
Vericiguat wurde in der Zulassungsstudie VICTORIA (1) bei Hochrisiko-Patienten (n = 5050) mit chronischer symptomatischer Herzinsuffizienz und reduzierter linksventrikulärer Ejektionsfraktion (LVEF < 45 %) untersucht. Die Patienten wurden 1:1 zu Vericiguat oder Placebo randomisiert, zusätzlich zu einer medikamentösen Begleittherapie der Herzinsuffizienz. Die Studie VICTORIA wurde doppelblind und multizentrisch durchgeführt. Die Studiendauer lag ereignisgesteuert bei knapp elf Monaten.
Eingeschlossen wurden Patienten, die aufgrund einer Dekompensation der Herzinsuffizienz innerhalb der letzten sechs Monate hospitalisiert wurden oder innerhalb der letzten drei Monate ambulant intravenös Diuretika erhielten. Ein akutes kardiales Ereignis oder eine koronare Revaskularisation musste mindestens 60 Tage zurück liegen. Patienten mit einem systolischen Blutdruck < 100 mmHg oder einer symptomatischen Hypotonie waren von der Studienteilnahme ausgeschlossen. Zudem stellte die Medikation mit langwirksamen Nitraten oder Phosphodiesterasehemmern (beispielsweise Sildenafil) ein Ausschlusskriterium dar.
Etwa ein Drittel der Patienten kam aus Osteuropa und 18 % aus Westeuropa. Das Durchschnittsalter lag bei 67 Jahren, 24 % der Studienteilnehmer waren Frauen. Die Patienten wiesen in der Mehrzahl prognostisch ungünstige Faktoren auf (mittlere LVEF 29 % medianer NT-proBNP-Wert 2816 pg/ml, Vorhofflimmern oder -flattern bei 53 % der Patienten). Knapp 60 % der Patienten waren bei Studienbeginn im NYHA(New York Heart Association)-Stadium II, 40 % im NYHA-Stadium III. Nur sehr wenige Patienten (n = 66; 1,3 %) mit NYHA-Stadium IV wurden in die Studie eingeschlossen. 28 % hatten einen implantierten Cardioverter-Defibrillator (ICD).
Die letzte Dekompensation lag im Median 32 Tage zurück und war überwiegend (84 %) stationär behandelt worden. Entsprechend den Ein- und Ausschlusskriterien war eine Randomisierung bereits 24 Stunden nach Beendigung der intravenösen Therapie möglich. Diese Zeitspanne erscheint zu kurz für eine Optimierung der oralen Diuretikatherapie. Es ist deshalb anzunehmen, dass der Volumenstatus zu Studienbeginn nicht bei allen Patienten stabilisiert war. Weder der Originalpublikation noch dem EPAR sind Angaben zur Intensität der Diuretikatherapie zu entnehmen.
Insgesamt entspricht die Begleittherapie der Herzinsuffizienz nicht dem Versorgungsstandard in Deutschland (2). In der Studie VICTORIA erhielten nur 60 % der Studienteilnehmer eine Dreifachtherapie (ACE-Hemmer bzw. ARB 73 %, Betablocker 93 % und MRA 70 %) und lediglich bei 15 % war eine Eskalation der Basistherapie mit Sacubitril/Valsartan erfolgt. SGLT2-Inhibitoren wurden kaum eingesetzt (3 % der Patienten). Die Gründe für die Nichtbehandlung sind unzureichend beschrieben; am häufigsten wurde bei allen Substanzen als Begründung die „Präferenz des Studienteilnehmers oder Arztes“ angegeben. Zudem fehlen Informationen zur Anpassung der Begleittherapie im Studienverlauf. So bleibt unklar, ob im Verlauf relevante Unterschiede in der Begleittherapie zwischen den Studienarmen auftraten.
Die Startdosis von Vericiguat lag bei 2,5 mg/Tag. Die Dosis wurde in Abständen von zwei Wochen verdoppelt, wenn der systolische Blutdruck mindestens 100 mmHg betrug. Knapp ein Drittel der Patienten erreichte die Maximaldosis von 10 mg/Tag Vericiguat. Im Studienprotokoll ist beschrieben, bei welchen Blutdruckwerten die Studienmedikation pausiert werden sollte, nicht aber, wann diese dauerhaft ausgesetzt werden musste. Die Abbruchrate der Studienmedikation war in beiden Armen gleichermaßen hoch mit je 23 % der Patienten.
Primärer Endpunkt der Studie VICTORIA war die Kombination aus kardiovaskulärem Tod oder Hospitalisierung aufgrund von Herzinsuffizienz. Die absolute Risikoreduktion durch Vericiguat betrug 3 %, entsprechend einer Number needed to treat von 24 (bezogen auf eine Behandlungsdauer über ein Jahr). Diese Risikoreduktion beruhte vor allem auf einer verringerten Häufigkeit von Hospitalisierungen aufgrund von Herzinsuffizienz. Kardiovaskuläre Todesfälle und Gesamtmortalität waren unter Vericiguat nicht signifikant reduziert (siehe Tabelle 1).
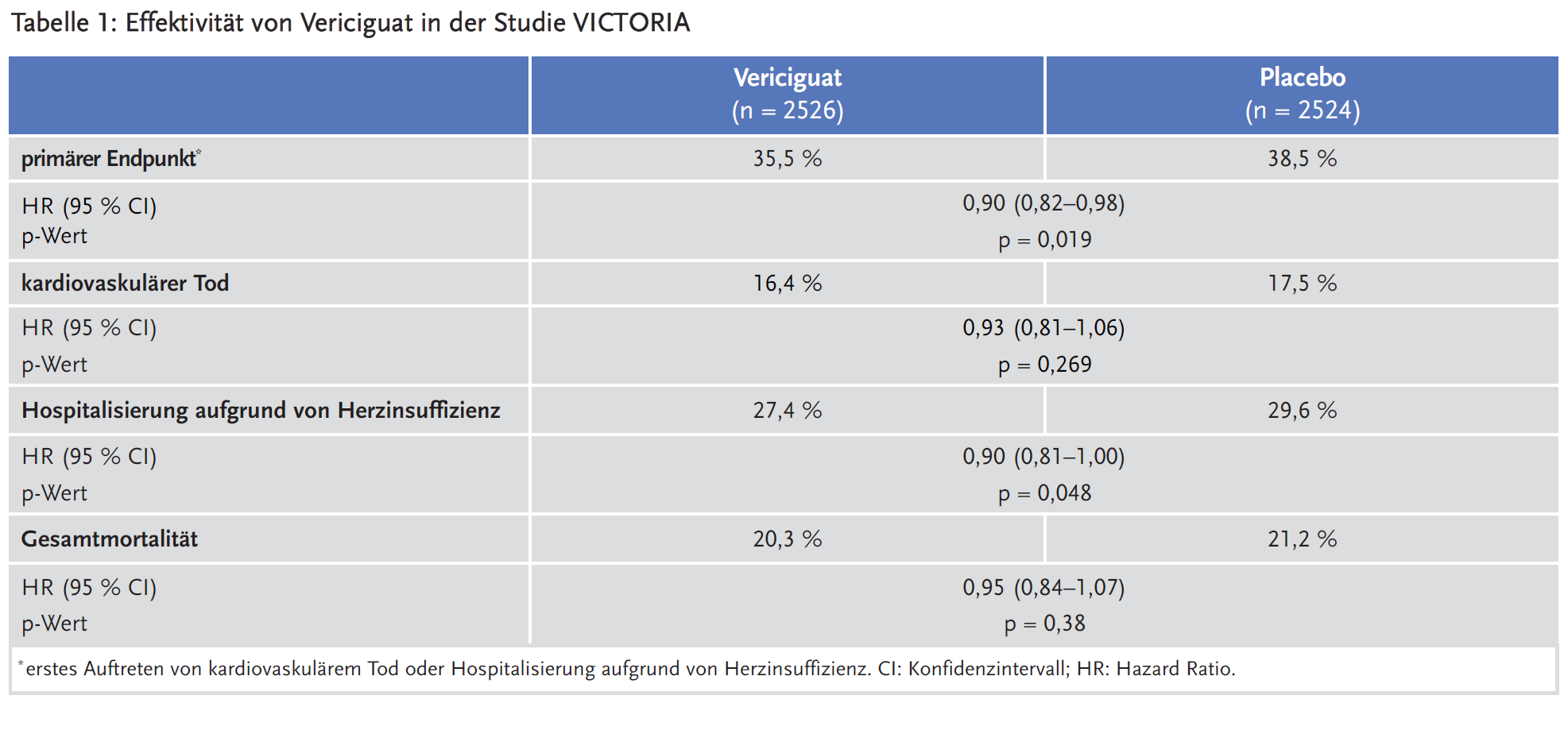
Post-hoc durchgeführte multivariate Analysen zeigten eine signifikante Effektmodifikation bei hohen NT-proBNP Ausgangswerten (4. Quartile NT-proBNP entsprechend NT-proBNP > 5314 pg/ml bei Studienbeginn: Hazard Ratio [HR] 1,16; 95 % Konfidenzintervall [CI] 0,99–1,35; p für Interaktion = 0,001). Nach Einschätzung der EMA entspricht diese Subgruppe klinisch instabilen Patienten, die eine weitere Optimierung der Herzinsuffizienztherapie, insbesondere durch Diuretika, benötigen. Die Zulassung wurde deshalb auf Patienten eingeschränkt, die „nach einem kürzlich aufgetretenen Dekompensationsereignis […] stabilisiert wurden“, wobei die Fachinformation darauf hinweist, dass diese Optimierung insbesondere bei Patienten mit sehr hohen NT-proBNP-Spiegeln erfolgen sollte (3). Unsicherheiten zur Effektivität bestehen auch für Patienten mit einem Alter ≥ 75 Jahren oder einer stark eingeschränkten Nierenfunktion, ohne dass hier allerdings ein signifikanter Interaktionstest vorliegt.
Ausgewählte Nebenwirkungen
Eine symptomatische Hypotonie trat numerisch häufiger unter Vericiguat auf als unter Placebo (9,1 % vs. 7,9 %; p = 0,12). Synkopen waren in beiden Armen etwa gleich häufig (4,0 % vs. 3,5 %). Im Studienverlauf entwickelten mehr Patienten unter Vericiguat eine Anämie als unter Placebo (7,6 % vs. 5,7 %). Auch schwerwiegende Anämien waren unter Vericiguat häufiger (1,6 % vs. 0,9 %). Laut EMA ist der pathophysiologische Mechanismus gehäufter Anämien unter Vericiguat unklar. Allerdings fiel dieses Risiko bereits früher in dieser Wirkstoffklasse auf: Unter Riociguat traten im Vergleich zur Kontrolle viermal so viele Anämien auf (7,8 % vs. 1,9 %). Hier führte die EMA die gehäuften Anämien auf eine Hämodilutation durch die cGMP-bedingte Vasodilatation zurück (4). Für diese Hypothese spricht, dass Blutungsereignisse unter Riociguat und Vericiguat nicht gehäuft auftraten.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Vericiguat kann eine vorbestehende Hypotonie verstärken. Vericiguat sollte deshalb nicht begonnen werden bei Patienten mit einer symptomatischen Hypotonie oder einem systolischen Blutdruck < 100 mmHg. Tritt eine Hypotonie unter der Behandlung mit Vericiguat auf, sollte Vericiguat pausiert werden, wenn der Patient symptomatisch ist oder der systolische Blutdruck unter 90 mmHg sinkt.
- In der Zulassungsstudie war die Einnahme von Phosphodiesterase-Hemmern nicht erlaubt. Bei gesunden Probanden führte die gleichzeitige Anwendung von Phosphodiesterase-Hemmern und Vericiguat zu einer zusätzlichen Senkung des Blutdrucks von etwa 5 mmHg. Von einer kombinierten Gabe von Phosphodiesterase-Hemmern und Vericiguat wird deshalb abgeraten.
- Patienten mit terminaler Niereninsuffizienz (GFR < 15 ml/min/1,73 m2) waren aus der Zulassungsstudie ausgeschlossen. Die Behandlung mit Vericiguat wird deshalb bei terminaler Niereninsuffizienz nicht empfohlen. Bei Patienten mit schwerer Niereninsuffizienz (GFR 15–29 ml/min/1,73m2) empfiehlt sich eine vorsichtige Dosistitration, da in einer pharmakokinetischen Studie diese Patienten eine 2,2-fach erhöhte Exposition von Vericiguat aufwiesen.

Weiterführende Informationen
Das IQWiG wurde am 15.09.2021 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Verquvo®, erschienen am 27. Juli 2021. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Literatur
- Armstrong PW, Pieske B, Anstrom KJ et al.: Vericiguat in patients with heart failure and reduced ejection fraction. N Engl J Med 2020; 382: 1883-1893.
- Nationale Versorgungsleitlinie: Chronische Herzinsuffizienz – Langfassung: www.leitlinien.de/themen/herzinsuffizienz (letzter Zugriff: 25. November 2021). Ärztliches Zentrum für Qualität in der Medizin (ÄZQ); 3. Auflage, Version 2; AWMF-Reg-Nr.: nvl-006. Oktober 2019.
- Bayer Vital: Fachinformation Verquvo® Filmtabletten. Stand: Juli 2021.
- European Medicines Agency (EMA): Adempas® – Riociguat: European Public Assessment Report (EPAR) – Assessment Report: www.ema.europa.eu/en/documents/assessment-report/adempas-epar-public-assessment-report_en.pdf (letzter Zugriff: 25. November 2021). London, Stand: 23. Januar 2014.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 14. Dezember 2021 vorab online veröffentlicht.