Rote-Hand-Briefe und Informationsbriefe – der Weg von der Entstehung bis zur Versendung an den Empfänger
Rote-Hand- und Informationsbriefe sollen Ärzte und Apotheker und gegebenenfalls andere Fachkreise schnell über Arzneimittelrisiken und deren Minimierung informieren. Auf europäischer Ebene sind sie unter dem Begriff Direct Healthcare Professional Communication (DHPC) oder umgangssprachlich als Dear Doctor Letter bekannt.
Warum werden Rote-Hand-Briefe und Informationsbriefe versendet?
Zum Zeitpunkt der Zulassung ist das Sicherheitsprofil von Arzneimitteln noch nicht vollständig bekannt. Nach der Zulassung und Markteinführung werden sie von vielen Patienten angewendet, wodurch neue Sicherheitsaspekte bekannt werden, wie Nebenwirkungen, Wechselwirkungen, Kontraindikationen, Fehlgebrauch, Anwendung bei kritischen Patientengruppen und andere Arzneimittelrisiken. Oft werden die einzelnen Arzneimittel oder ganze Stoffgruppen im Rahmen eines europäischen Verfahrens untersucht, z. B. Risikobewertungsverfahren (Fluorchinolon-Antibiotika: schwerwiegende und anhaltende, die Lebensqualität beeinträchtigende und möglicherweise irreversible Nebenwirkungen) (1) oder Signalverfahren (Sildenafil-haltige Arzneimittel: keine Anwendung zur Behandlung der intrauterinen Wachstumsrestriktion) (2).
Einige der so gewonnenen neuen Erkenntnisse erfordern ein unmittelbares Handeln für den Arzt und/oder Apotheker und müssen ihnen schnellstmöglich mitgeteilt werden. Da die Aufnahme in die Fach- und Gebrauchsinformation des Arzneimittels eine gewisse Zeit in Anspruch nimmt und entsprechende Änderungen erst mit einer Zeitverzögerung zur Kenntnis genommen werden, kann es daher erforderlich sein, diese wichtigen Informationen schnell den Fachkreisen mittels Rote-Hand-Brief mitzuteilen. Gemäß § 11a Abs. 2 Arzneimittelgesetz (AMG) ist der pharmazeutische Unternehmer verpflichtet, die Fachkreise über Änderungen, die therapierelevant sind, sowie über neu erkannte Arzneimittelrisiken zu informieren. Solche Risikoinformationen können z.B. eine neue Kontraindikation, neue Warnhinweise oder ein Rückruf aufgrund eines Qualitätsmangels eines Präparates sein.
Pharmazeutische Unternehmer (pU) müssen geplante Risikoinformationen zu Arzneimitteln wie Rote-Hand-Briefe mit den zuständigen nationalen Behörden im Hinblick auf Inhalt und Adressatenkreis abstimmen (§ 63b Abs. 3 AMG und Artikel 106a Abs. 1 der Richtlinie 2001/83/EG). Die Informationen müssen objektiv dargestellt werden und dürfen weder irreführend formuliert sein noch Werbung enthalten. Der Versand selbst erfolgt aber in Eigenverantwortung des pU, bei mehreren beteiligten pharmazeutischen pU in der Regel koordiniert durch die Verbände der pharmazeutischen Industrie.
Was ist der Unterschied zwischen einem Rote-Hand-Brief und einem Informationsbrief?
Ein Rote-Hand-Brief soll die Fachkreise rasch über neue therapierelevante Aspekte informieren, die noch nicht in der Fachinformation enthalten sind. Dies ist z. B. notwendig, wenn die Information eine Änderung des Verschreibungsverhaltens der Ärzte erforderlich macht und/oder einen unmittelbaren, therapiebeeinflussenden Handlungsbedarf erfordert. Dies können eine neue Kontraindikation, ein Warnhinweis, der Rückruf eines Präparates/einer Charge aufgrund eines medizinischen Risikos, eines erheblichen Qualitätsmangels, oder der Gefahr einer Verwechslung sein. Rote-Hand-Briefe werden auch zur Erinnerung an die Einhaltung wichtiger bestehender Kontraindikationen und Warnhinweise verwendet, wenn z. B. Erkenntnisse vorliegen, dass solche Vorgaben bei der Verordnung nicht genügend Beachtung finden.
Dagegen informiert ein Informationsbrief die Fachkreise beispielsweise über eine Erweiterung der Zulassung (z. B. Anwendungserweiterung) oder über bestimmte qualitative Aspekte (z. B. geänderte farbliche Markierung auf der Primärverpackung zur Vermeidung von Verwechslung). Diese Informationen helfen den Fachkreisen, die Anwendung des Arzneimittels sicherer zu gestalten. Sie haben keinen unmittelbaren Einfluss auf die Therapie und werden daher nicht mit dem Symbol der Roten Hand gekennzeichnet. Die Informationsbriefe werden in der Regel auch mit der zuständigen Bundesoberbehörde (BfArM oder PEI) oder mit der zuständigen Landesbehörde abgestimmt. Ein Informationsbrief kann aber grundsätzlich auch eigenverantwortlich durch den pharmazeutischen Unternehmer ohne Abstimmung mit den Behörden versendet werden. Analog zu den Rote-Hand-Briefen sollen die Informationsbriefe weder Werbung noch irreführende Aussagen enthalten.
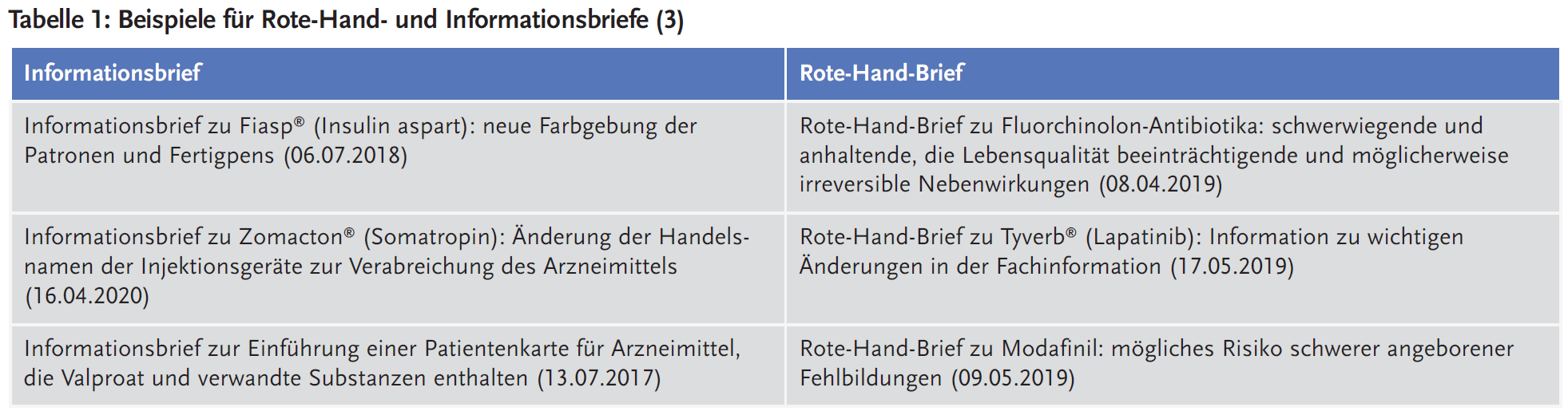
In Tabelle 1 sind Beispiele von Rote-Hand- und Informationsbriefen zum direkten Vergleich dargestellt.
Wer bekommt die Rote-Hand-Briefe und Informationsbriefe?
Der überwiegende Teil der Rote-Hand- und Informationsbriefe resultiert aus europäischen Risikobewertungs- oder Signalverfahren, in denen Risiken evaluiert und Maßnahmen zu deren Minimierung beschlossen werden. Wenn die neuen Risikoinformationen bzw. die verabschiedeten Maßnahmen in Form einer DHPC schnell an die Fachkreise kommuniziert werden müssen, wird in der Regel festgelegt, für welche Zielgruppen eine DHPC jeweils relevant ist. Dies können Ärzte der verschiedensten Fachrichtungen sein, Apotheker, aber auch Fachgesellschaften, Pflegepersonal etc. Die Arzneimittelkommission der Deutschen Apotheker (AMK) und die Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) werden in Deutschland routinemäßig informiert. Die Versendung eines Rote-Hand-Briefes kann aber auch rein national durch die zuständige Bundesoberbehörde angeordnet werden.
Wer versendet die Rote-Hand-Briefe und Informationsbriefe an die Fachkreise?
Rote-Hand-Briefe und Informationsbriefe werden durch die pharmazeutischen Unternehmer eigenverantwortlich als risikominimierende Maßnahme versendet. Sie sind definiert in § 14 der Kodizes der Vereine „Freiwillige Selbstkontrolle für die Arzneimittelindustrie e.V.“ (FSA) und „Arzneimittel und Kooperation im Gesundheitswesen e.V.“ (AKG e.V.). Die Bildmarke des Symbols der Roten Hand wurde im Jahr 2000 von Bundesverband der Pharmazeutischen Industrie e.V. rechtlich geschützt. Der Inhalt und der Empfängerkreis werden je nach Zuständigkeit bei Humanarzneimitteln mit dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) oder Paul-Ehrlich-Institut (PEI) oder bei Qualitätsmängeln mit der zuständigen Landesbehörde abgestimmt. Die pharmazeutischen Unternehmer beauftragen nach Freigabe durch die Behörde die entsprechenden Druckereien und Distributoren mit dem Versand, der zurzeit ausschließlich per Post erfolgt.
Ist die Information im Rote-Hand-Brief/ Informationsbrief verbindlich?
Die im Rote-Hand-Brief enthaltenen Informationen stellen jeweils den neuesten wissenschaftlichen Kenntnisstand dar, über den die Fachkreise informiert sein müssen. Es ist aus behördlicher Sicht entscheidend, dass die Fachkreise diese Risikokommunikation als ein wichtiges Instrument zur Minderung der Anwendungsrisiken von Arzneimitteln wahrnehmen und nicht irrtümlich als postalische Routinesendung mit nur geringer Bedeutung fehlinterpretieren. Die dargestellten Informationen finden entsprechend auch Eingang in die Fach- und Gebrauchsinformation der betroffenen Arzneimittel und sind in der ärztlichen Praxis daher sorgfältig zu berücksichtigen.
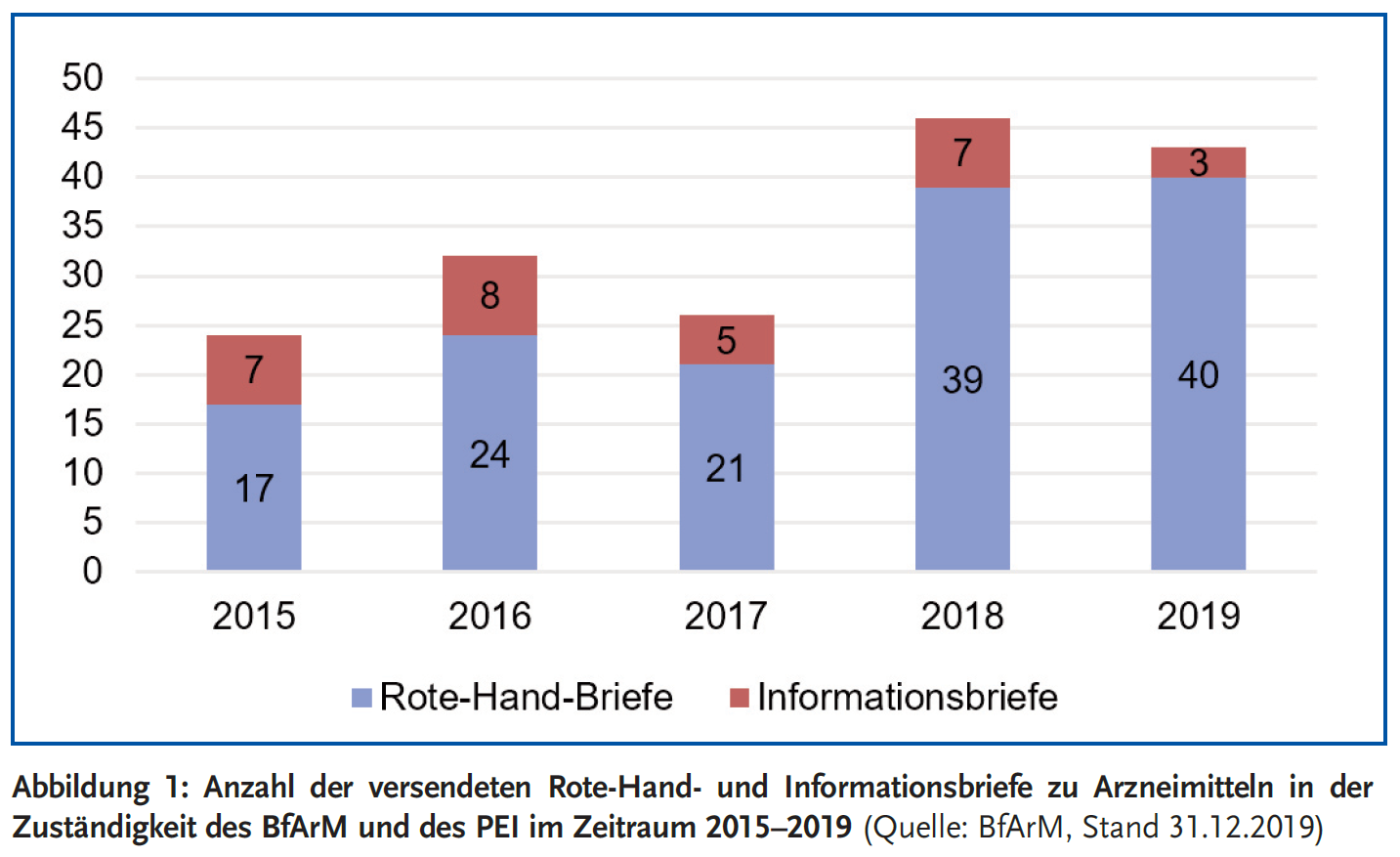
Die Abbildung 1 zeigt die Anzahl der versendeten Rote-Hand- bzw. Informationsbriefe im Zeitraum 2015–2019 in der Zuständigkeit des BfArM und des PEI.
Wann bekomme ich einen Rote-Hand- oder Informationsbrief persönlich?
Nicht jeder Arzt bekommt jeden Rote-Hand-Brief und Informationsbrief persönlich per Post. Dies richtet sich u. a. nach der Fachrichtung und Anwendung des jeweiligen betroffenen Arzneimittels. Wenn sich z. B. ein Rote-Hand-Brief auf ein Risiko bezieht, das ein Arzneimittel mit gynäkologischer Indikation betrifft, wird dieser hauptsächlich an Fachärzte für Gynäkologie und Geburtshilfe (sowohl niedergelassene als auch in der Klinik praktizierende Ärzte) versendet, gegebenenfalls auch an Hebammen und Geburtshelfer sowie an Ärzte anderer Fachrichtungen, sofern davon auszugehen ist, dass diese die betroffenen Arzneimittel ebenfalls anwenden bzw. verordnen könnten. Im genannten Beispiel würden Fachärzte für z. B. Augenheilkunde oder Orthopädie nicht zum Adressatenkreis gehören.
Kann ich eine elektronische Version anstatt eines Briefes per Post bekommen?
In Deutschland wird der Rote-Hand-Brief derzeit postalisch an die definierten Zielgruppen (Ärzte, Apotheker, medizinische Fachkreise) verschickt und auf den jeweiligen Websites der Zulassungsinhaber, der Bundesoberbehörden BfArM und PEI, sowie der Arzneimittelkommissionen veröffentlicht. Durch diese Zustellung soll sichergestellt werden, dass der Empfänger bestmöglich erreicht wird und Kenntnis von der Information erhält.
Es wird jedoch aktuell intensiv diskutiert, ob eine direkte elektronische Zustellung zeitgemäßer, kostengünstiger und vor allem schneller ist.
In der Vergangenheit haben insbesondere Ärzte berichtet, dass sie Rote-Hand-Briefe nicht immer zeitnah erhalten hatten. Daher ist zu prüfen, inwieweit technische Weiterentwicklungen genutzt werden können, um flächendeckend alle Adressaten des jeweiligen Rote-Hand- oder Informationsbriefes schnell und zuverlässig zu erreichen. Eine Möglichkeit ist die Implementierung von Rote-Hand- und Informationsbriefen in die Apotheken- und Praxissoftware, die in ein großes Potenzial hat, da die Informationen den Fachkreisen papierlos zur Verfügung gestellt und dem Arzneimittel im System direkt zugeordnet werden. Es müsste allerdings dabei sichergestellt werden, dass die Anbieter von Arzneimittel-Verordnungssoftware Rote-Hand- bzw. Informationsbriefe verlässlich und kurzfristig nach der Veröffentlichung in die Software implementieren.
Eine andere mögliche elektronische Alternative wäre die Versendung an die Fachverbände in deren Funktion als Multiplikatoren zur Weiterverteilung an die einzelnen Ärzte der jeweiligen Fachrichtungen. Dies betrifft insbesondere Fachärzte in der Klinik. Eine Versendung per E-Mail oder die Nutzung einer App wurden in der Vergangenheit ebenfalls diskutiert, aber nicht als die beste aller denkbaren Alternativen weiterverfolgt.
Wo finde ich alle Rote-Hand- Briefe und Informationsbriefe?
Alle Rote-Hand-Briefe und Informationsbriefe sind elektronisch verfügbar. Sie sind auf der Webseite der AkdÄ veröffentlicht: https://www.akdae.de/Arzneimittelsicherheit/RHB/. Eine elektronische Benachrichtigung durch die AkdÄ ist über ein kostenloses Abonnement des Newsletters „Drug Safety Mail“ möglich.
Alle Rote-Hand-Briefe und Informationsbriefe zu Arzneimitteln in der BfArM-Zuständigkeit sowie Qualitätsmängel betreffend (Zuständigkeit Landesbehörden) sind auf der BfArM-Homepage abrufbar unter:
https://www.bfarm.de/DE/Arzneimittel/Pharmakovigilanz/Risikoinformationen/Rote-Hand-Briefe_Informationsbriefe/_functions/RI_rhb_Filtersuche_Formular.html.
Dort besteht auch die Möglichkeit einen RSS-Feed, den Newsletter oder auch das Bulletin zur Arzneimittelsicherheit zu abonnieren. Alle Rote-Hand-Briefe zu Arzneimitteln in der Zuständigkeit des Paul-Ehrlich- Instituts (PEI) sind verfügbar unter:
https://www.pei.de/DE/newsroom/veroffentlichungen-arzneimittel/rote-hand-briefe/rote-hand-briefe-node.html.
Interessenkonflikte
Ein Interessenkonflikt wird von der Autorin verneint.
Literatur
- www.ema.europa.eu/en/events/pharmacovigilance-risk-assessment-committee-prac-11-14-june-2018
- www.ema.europa.eu/en/documents/minutes/minutes-prac-meeting-3-6-september-2018_en.pdf
- www.bfarm.de/DE/Arzneimittel/Pharmakovigilanz/Risikoinformationen/Rote-Hand-Briefe_Informationsbriefe/_functions/RI_rhb_Filtersuche_Formular.html
Hinweise
Zuständige Bundesoberbehörden in Deutschland sind nach § 77 AMG:
- Paul-Ehrlich-Institut (PEI): für Sera, Impfstoffe, Blutzubereitungen, Gewebezubereitungen, Gewebe, Allergene, Arzneimittel für neuartige Therapien (ATMP), xenogene Arzneimittel und gentechnisch hergestellte Blutbestandteile (www.pei.de).
- Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM): für alle anderen humanen Arzneimittel bzw. für alle Medizinprodukte (www.bfarm.de).
- Aufgaben der Bundesoberbehörden: Zulassung und Registrierung von Arzneimitteln, Erfassung und Auswertung von Arzneimittelrisiken und Koordinierung der zu ergreifenden Maßnahmen, Genehmigung klinischer Prüfungen und staatliche Chargenprüfung bei Sera, Impfstoffen, Allergenen (PEI).
Zuständige Behörden der Länder:
Die Überwachung des Verkehrs mit Arzneimitteln, also von Herstellung, Einfuhr, Vertrieb und klinischen Prüfungen von Arzneimitteln sowie die Überwachung der pharmazeutischen Unternehmer, des Groß- und Einzelhandels und der Apotheken obliegt den Behörden der Länder (www.zlg.de).
vorab online
Dieser Artikel wurde am 9. Oktober 2020 vorab online veröffentlicht.