Romosozumab (Evenity®) ▼
Zugelassene Indikation und Wirkmechanismus
Romosozumab (Evenity®) ist für die Behandlung der manifesten Osteoporose bei postmenopausalen Frauen mit deutlich erhöhtem Frakturrisiko zugelassen. Romosozumab ist ein humanisierter monoklonaler Antikörper (IgG2). Romosozumab hemmt Sklerostin, einen Botenstoff der Osteozyten, der als negativer Regulator der Knochenbildung fungiert. Dadurch wird aufgrund der Aktivierung von Saumzellen (Cellulae vestientes osseorum), der gesteigerten Knochenmatrixproduktion durch Osteoblasten und Rekrutierung von Osteoprogenitorzellen der Knochenaufbau gestärkt. Zusätzlich führt Romosozumab zu Veränderungen bei der Expression von Osteoklastenmediatoren und hemmt dadurch den Knochenabbau. Der duale Wirkmechanismus – verstärkter Knochenaufbau und gehemmter Knochenabbau – soll zu einem raschen Anstieg der trabekulären und kortikalen Knochenmasse sowie zu einer Verbesserung der Knochenstruktur und der Festigkeit führen.
Markteinführung
Romosozumab (Evenity®) ist seit dem 15. März 2020 in dieser Indikation auf dem deutschen Markt verfügbar.
Bewertung
Romosozumab ist nach Teriparatid der zweite osteoanabole Wirkstoff zur Behandlung der postmenopausalen manifesten Osteoporose bei deutlich erhöhtem Frakturrisiko. Die osteoanabole Wirkung hält allerdings nur einige Monate an. Eine einjährige Behandlung mit Romosozumab senkte in den beiden Zulassungsstudien das Risiko neuer radiologischer Wirbelkörperfrakturen nach 12 und 24 Monaten im Vergleich zu Placebo (FRAME: 0,5 % vs. 1,8 % über 12 Monate und 0,7 % vs. 2,5 % über 24 Monate) und im Vergleich zu Alendronsäure (ARCH: 3,2 % vs. 5,0 % über 12 Monate und 4,1 % vs. 8,0 % über 24 Monate). In der Studie ARCH reduzierte Romosozumab auch das Risiko klinischer Frakturen (vertebrale und nicht vertebrale) im Vergleich zu Alendronsäure nach 12 Monaten (3,9 % vs. 5,4 %) und im Vergleich zu Alendronsäure nach 33 Monaten (9,7 % vs. 13,0 %). Die Wirkung auf nicht vertebrale Frakturen und Hüftfrakturen erreichte in den Studien keine statistische Signifikanz bis Monat 24. In der ARCH-Studie war Romosozumab nach 33 Monaten Alendronsäure statistisch signifikant überlegen bezüglich der Reduktion des Risikos für nicht vertebralen Frakturen (10,6 % vs. 8,7 %) und Hüftfrakturen (3,2 % vs. 2,0 %). Die sequenzielle Therapie mit Romosozumab und anschließend Alendronsäure oral erscheint daher sowohl bei vertebralen als auch bei nicht vertebralen Frakturen im Vergleich zu der derzeitigen Standardbehandlung mit Alendronsäure in dieser Patientenpopulation besser wirksam. Für einen direkten Vergleich von Romosozumab mit anderen antiresorptiven Arzneimitteln wie Zolendronsäure und Denosumab liegen keine Daten vor. Zudem gibt es Beobachtungen, die eine Anwendung von Romosozumab bei TNF-α-vermittelten Erkrankungen (z. B. rheumatoide Arthritis, M. Crohn) oder bei immunkompromitierten Patienten (z. B. nach Transplantionen, mit Glukokortikoid-Dauertherapie) ohne vorliegende weitere Studien ungünstig erscheinen lassen.1
In der Zulassungsstudie ARCH zeigte sich zudem ein Anstieg kardiovaskulärer Ereignisse: Im ersten Jahr erlitten 2,5 % der Patientinnen unter Romosozumab ein schwerwiegendes kardiovaskuläres Ereignis (Herzinfarkt, Schlaganfall) vs. 1,9 % unter Alendronsäure. Diese Imbalance zeigte sich auch in der offenen Verlängerungsphase bis Monat 24, aber nicht in der FRAME-Studie. Um diesem Risiko Rechnung zu tragen, wurde die Indikation von Romosozumab auf Frauen ohne Herzinfarkte und Schlaganfälle in der Anamnese eingeschränkt.
Romosozumab ist daher als Ausweichbehandlung bei postmenopausalen Frauen mit manifester Osteoporose und deutlich erhöhtem Frakturrisiko und ohne kardio- oder zerebrovaskuläre Risiken zu sehen, nicht aber bei renaler Osteopathie, nierentransplantierten oder dialysepflichtigen Patienten, sowie TNF-α-vermittelten Erkrankungen oder immunkompromittierten Patienten. Zum Erhalt einer günstigen Wirkung auf die Knochenmasse ist nach Beendigung einer Romosozumab-Therapie eine Nachbehandlung mit einem Bisphosphonat zweckmäßig, da ebenfalls wie auch bei z. B. Teriparatid oder Denosumab innerhalb von Monaten mit einem Abfall der Wirkung dieses Antikörpers auf die Knochendichte nach Pausieren der monatlichen Romosozumab-Gabe zu rechnen ist.
Wirksamkeit in den Zulassungsstudien
Für die Zulassung von Romosozumab reichte der pharmazeutische Unternehmer drei multizentrische, randomisierte, doppelblinde Phase-III-Studien ein: zwei Studien an postmenopausalen Frauen (FRAME und ARCH) und eine Studie an Männern mit Osteoporose (BRIDGE). Zudem wurde eine unterstützende, offene Studie (STRUCTURE) eingereicht, die Romosozumab mit Teriparatid bei postmenopausalen Frauen mit manifester Osteoporose verglich, die von einer oralen Bisphosphonattherapie auf Romosozumab umgestellt wurden (Tabelle 1).
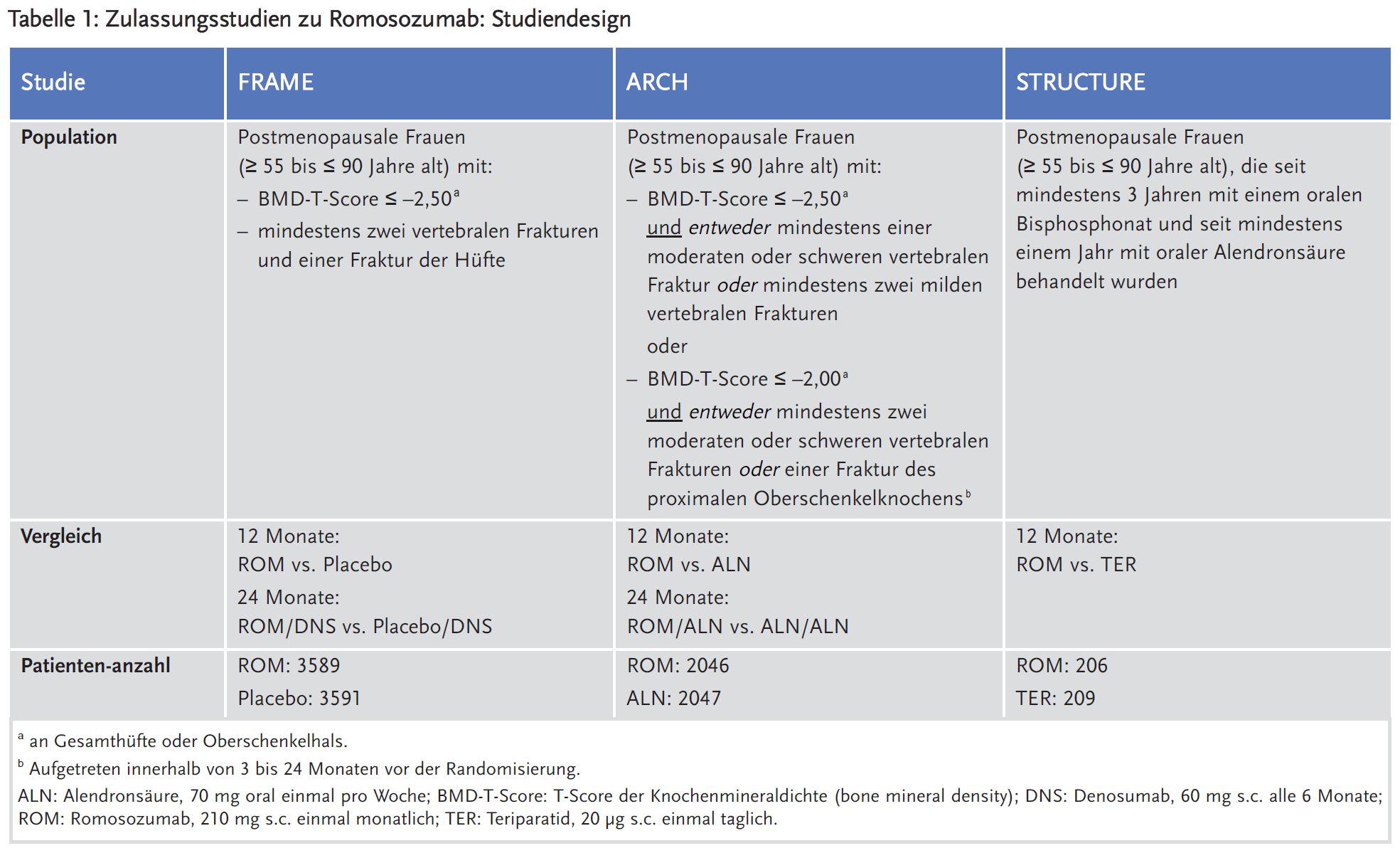
Die Studien FRAME und ARCH bestanden aus einer 12-monatigen Behandlungsphase und einer daran anschließenden, offenen, 24-monatigen Verlängerungsphase, in der die Patienten entweder mit Denosumab oder Alendronsäure behandelt wurden. Der primäre Wirksamkeitsendpunkt war die Inzidenz neuer vertebraler Frakturen bis einschließlich Monat 12 bzw. 24. Als sekundäre Endpunkte wurden u. a. erhoben: klinische Frakturen (alle symptomatischen Frakturen, einschließlich nicht vertebraler und schmerzhafter vertebraler Frakturen); nicht vertebrale Frakturen; Hüftfrakturen; typische osteoporosebedingte Frakturen (major osteoporotic fractures, MOF; Frakturen von Hüfte, Unterarm und Humerus sowie klinische vertebrale Frakturen).
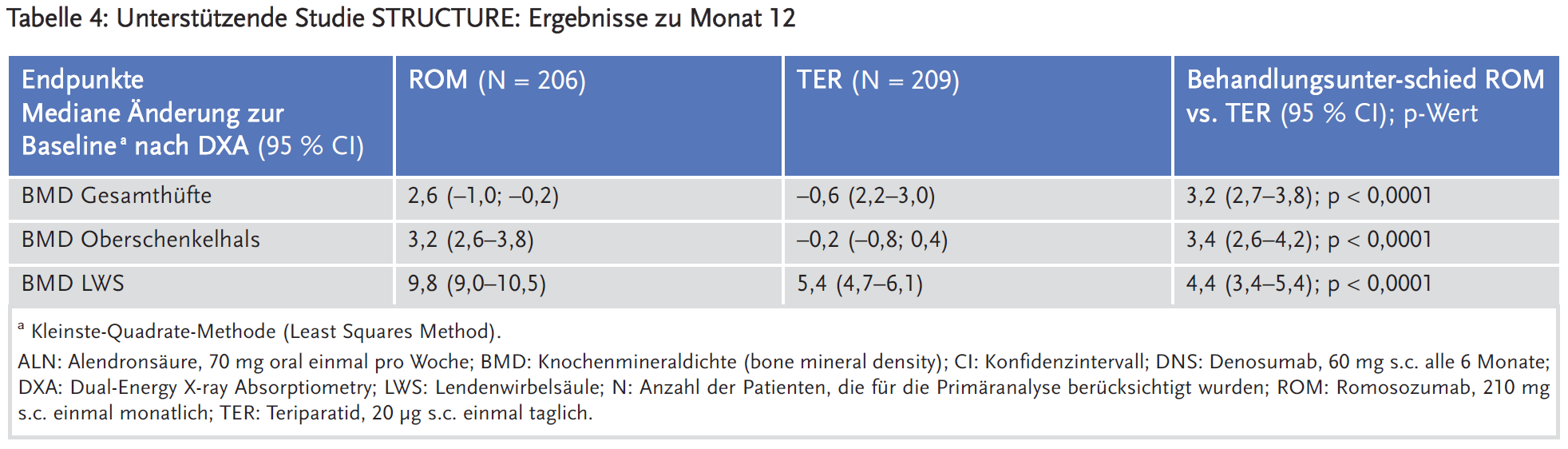
Die STRUCTURE-Studie schloss 415 Frauen ein (Tabelle 1). Der primäre Wirksamkeitsendpunkt war die prozentuale Änderung der Knochendichte der Gesamthüfte im Monat 12 gegenüber Baseline. Als sekundäre Endpunkte wurde erhoben u. a. die prozentuale Änderung der Knochendichte des proximalen Oberschenkelknochens und der Lendenwirbelsäule im Monat 12 gegenüber Baseline.
Die Ergebnisse zu den primären Wirksamkeitsendpunkten und zu ausgewählten sekundären Endpunkten zu Monat 12 und 24 sind für die Studien ARCH und FRAME in den Tabellen 2 und 3 und für die Studie STRUCTURE in Tabelle 4 dargestellt. In der ARCH-Studie wurde auch eine Primäranalyse durchgeführt, sobald alle Frauen den Besuchstermin in Monat 24 abgeschlossen hatten und bei mindestens 330 Frauen klinische Frakturereignisse bestätigt wurden, die nach einer medianen Beobachtungszeit von etwa 33 Monaten in der Studie auftraten.2
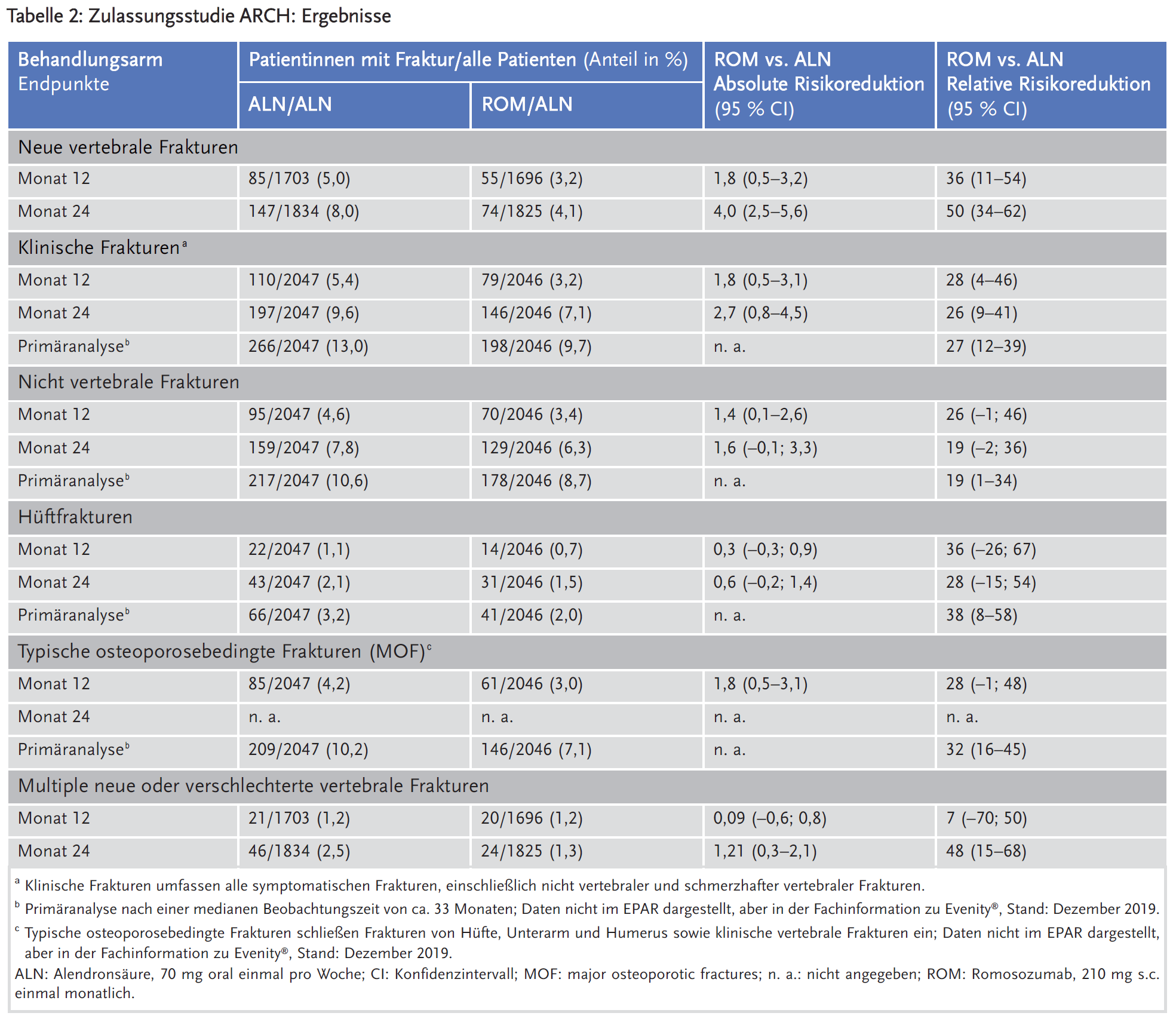
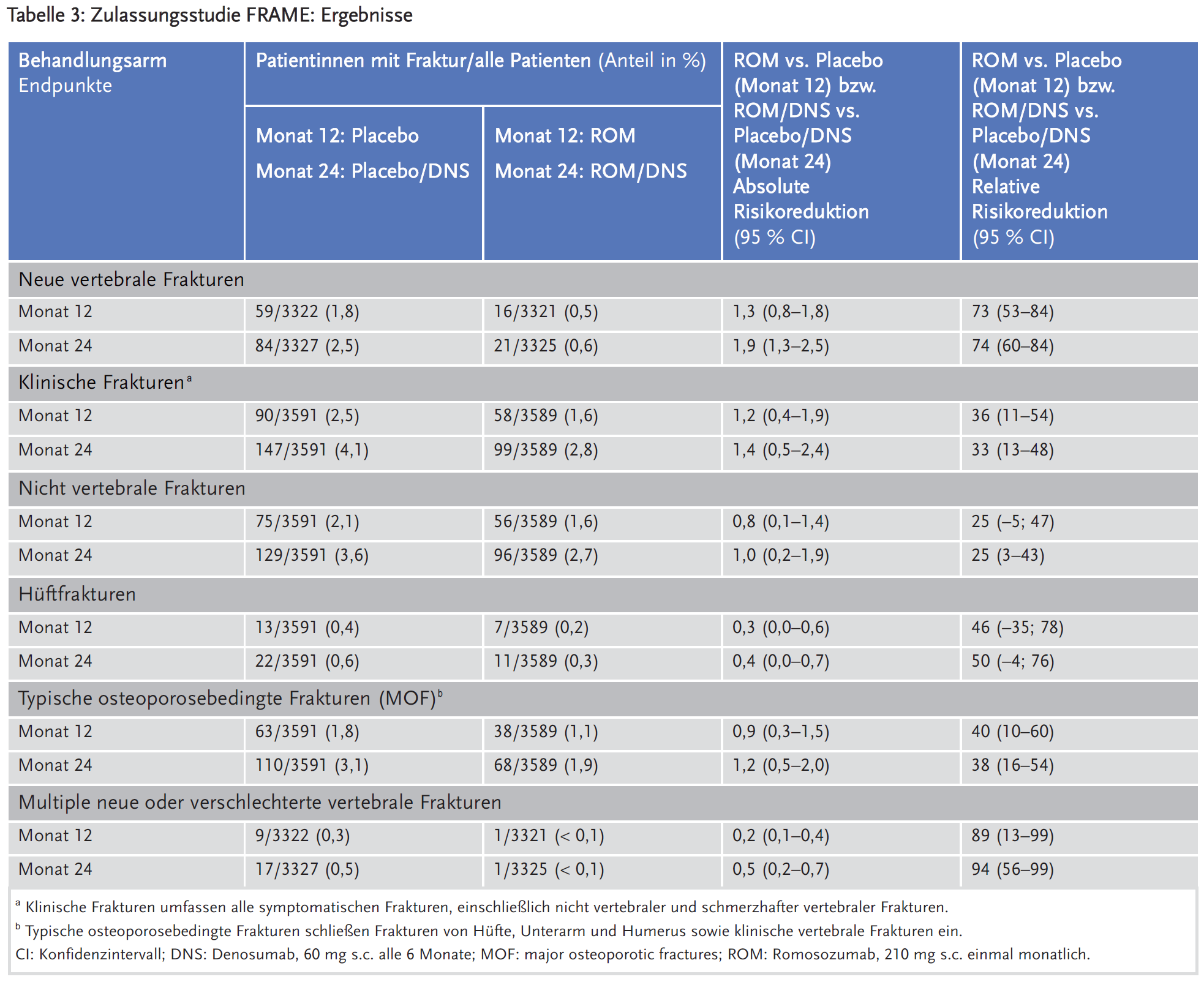
Eine einjährige Behandlung mit Romosozumab senkte in den beiden Zulassungsstudien das Risiko neuer radiologischer Wirbelkörperfrakturen nach 12 und 24 Monaten im Vergleich zu Placebo (FRAME: 0,5 % vs. 1,8 % über 12 Monate und 0,7 % vs. 2,5 % über 24 Monate) und im Vergleich zu Alendronsäure (ARCH: 3,2 % vs. 5,0 % über 12 Monate und 4,1 % vs. 8,0 % über 24 Monate).
In der ARCH-Studie reduzierte Romosozumab auch das Risiko klinischer Frakturen (vertebrale und nicht vertebrale) im Vergleich zu Alendronsäure nach 12 Monaten (3,9 % vs. 5,4 %) und im Vergleich zu Alendronsäure nach 33 Monaten (9,7 % vs. 13,0 %). Die Wirkung auf nicht vertebrale Frakturen und Hüftfrakturen (sekundäre Endpunkte) erreichte in den Studien keine statistische Signifikanz bis Monat 24. In der ARCH-Studie war Romosozumab zum Zeitpunkt der primären Analyse nach 33 Monaten Alendronsäure statistisch signifikant überlegen bezüglich der Reduktion des Risikos für nicht vertebrale Frakturen (10,6 % vs. 8,7 %) und Hüftfrakturen (3,2 % vs. 2,0 %).
Die sequenzielle Therapie mit Romosozumab und anschließend Alendronsäure oral scheint daher sowohl bei vertebralen als auch bei nicht vertebralen Frakturen einen höheren Effekt im Vergleich zu der derzeitigen Standardbehandlung zu erzielen. Für einen direkten Vergleich von Romosozumab mit anderen antiresorptiven Arzneimitteln wie Zolendronsäure und Denosumab liegen keine Daten vor.
In der Zulassungsstudie ARCH zeigte sich aber auch ein Anstieg kardiovaskulärer Ereignisse: Im ersten Jahr erlitten 2,5 % der Patientinnen unter Romosozumab ein schwerwiegendes kardiovaskuläres Ereignis (Herzinfarkt, Schlaganfall) vs. 1,9 % unter Alendronsäure. Schwere kardiale Komplikationen (major adverse cardiac event, MACE) traten bei 2,6 % der Patienten unter Romosozumab bzw. 1,6 % der Patienten unter Alendronsäure auf (HR 1,7; 95 % Konfidenzintervall [CI] 1,1–2,6). Diese Imbalance zeigte sich auch in der offenen Verlängerungsphase bis Monat 24, aber nicht in der FRAME-Studie (HR 1,1; 95 % CI 0,7–1,7).
Mögliche Erklärungen zum Pathomechanismus der beobachteten kardiovaskulären Ereignisse gehen von einer Schutzfunktion von Sklerostin in den Blutgefäßen aus, oder aber auch von einer gefäßprotektiven Wirkung von Alendronsäure, die zu einem scheinbaren Anstieg kardiovaskulärer Ereignisse geführt haben könnte. Um diesem Risiko Rechnung zu tragen, wurde die Indikation von Romosozumab auf Frauen ohne Herzinfarkte und Schlaganfälle in der Anamnese eingeschränkt, weil die EMA davon ausgeht, dass dadurch das Risiko kardiovaskulärer Nebenwirkungen verringert ist.
Ausgewählte Nebenwirkungen
Unter Romosozumab treten sehr häufig Nasopharyngitis (13,6 %) und Arthralgie (12,4 %) auf. Häufige Nebenwirkungen sind Überempfindlichkeitsreaktionen, Sinusitis, Kopfschmerzen, Nackenschmerzen, Muskelkrämpfe und Reaktionen an der Injektionsstelle (v. a. Schmerzen und Erytheme). Hypokalzämie, Urtikaria und Katarakt wurden gelegentlich berichtet.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Die empfohlene Dosis beträgt einmal monatlich 210 mg Romosozumab (als zwei subkutane Injektionen von je 105 mg) über einen Zeitraum von 12 Monaten. Die Injektion erfolgt in Bauch, Oberschenkel oder Oberarm, die zweite Injektion sollte unmittelbar nach der ersten erfolgen, jedoch an einer anderen Injektionsstelle.
- Die Patienten sollten vor und während der Behandlung ausreichend Kalzium und Vitamin D einnehmen.
- Romosozumab ist kontraindiziert bei Patienten mit Hypokalzämie. Vor Beginn der Therapie mit Romosozumab sollte der Kalziumspiegel bestimmt und eine Hypokalzämie behandelt werden. Die Patienten sollten auf Anzeichen und Symptome einer Hypokalzämie wie z. B. Muskelkrämpfe und/oder -spasmen, Parästhesie der Extremitäten, Gesichtszuckungen, Krampfanfälle und neuropsychiatrische Symptome überwacht werden.
- Nach Abschluss der Therapie mit Romosozumab ist die Umstellung auf eine antiresorptive Therapie angebracht, um den mit Romosozumab erzielten Nutzen über 12 Monate hinaus zu erhalten.
- Bei älteren Patienten und bei Patienten mit Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Bei Patienten mit schwerer Nierenfunktionsstörung (eGFR = 15 bis 29 ml/min/1,73 m2) oder bei Dialysepatienten sollte der Kalziumspiegel im Serum überwacht werden, da sie stärker gefährdet sind, eine Hypokalzämie zu entwickeln.
- In den Studien wurde unter Romosozumab im Vergleich zu Kontrollen ein Anstieg in der Häufigkeit von schweren kardiovaskulären Ereignissen (Myokardinfarkt und Schlaganfall) beobachtet. Romosozumab ist daher bei Patienten mit einem vorausgegangenen Myokardinfarkt oder Schlaganfall kontraindiziert.
- Bei der Entscheidung für den Einsatz von Romosozumab sollte das individuelle Frakturrisiko in den nächsten 12 Monaten sowie das individuelle kardiovaskuläre Risiko einbezogen werden unter Berücksichtigung folgender Risikofaktoren: bekannte kardiovaskuläre Erkrankung, Hypertonie, Hyperlipidämie, Diabetes mellitus, Rauchen, schwere Nierenfunktionsstörung, Alter.
- In klinischen Studien traten unter Romosozumab klinisch relevante Überempfindlichkeitsreaktionen auf, wie z. B. Angioödem, Erythema multiforme und Urtikaria. Wenn eine anaphylaktische oder andere klinisch relevante allergische Reaktion auftritt, muss eine geeignete Therapie eingeleitet und die Anwendung von Romosozumab eingestellt werden.
- Unter Romosozumab können Osteonekrosen des Kiefers auftreten. Risikofaktoren für die Entstehung einer Osteonekrose des Kiefers sollten berücksichtigt werden. Alle Patienten sollten zu guter Mundhygiene und routinemäßigen zahnärztlichen Untersuchungen angehalten werden und während der Behandlung mit Romosozumab sofort alle oralen Symptome wie Zahnmobilität, Schmerzen oder Schwellungen oder nicht heilende oder sezernierende Wunden melden.
Schulungsmaterial
Für einzelne Arzneimittel wird bereits bei der Zulassung angeordnet, dass das Arzneimittel nur unter Verwendung von Schulungsmaterialien in Verkehr gebracht werden darf. Das Schulungsmaterial dient dazu, die Wissensvermittlung zu optimieren und Hilfe bei der sicheren Anwendung des Arzneimittels zu geben, ggfs. unter Einbeziehung einer patientenbezogenen Ansprache. Das behördlich beauflagte und genehmigte Schulungsmaterial zu Romosozumab beinhaltet einen Leitfaden für Ärzte (https://www.pei.de/SharedDocs/schulungsmaterial/Evenity-Schulungsmaterial-Aerzte_Version-1_Leitfaden-Verschreiber.pdf?) sowie eine Patienteninformationskarte (https://www.pei.de/SharedDocs/schulungsmaterial/Evenity-Schulungsmaterial-Aerzte_Version-1_Patienteninformation.pdf?).

Weiterführende Informationen
Das IQWiG wurde am 15.03.2020 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Evenity®, erschienen am 24. Februar 2020. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Fußnoten
1Kasperk HC: Zweckmäßige Diagnostik und medikamentöse Therapie der Osteo- porose. AVP 2020; (47): 26-37.
2Informationen zur zweckmäßigen Diagnostik und medikamentösen Therapie der Osteoporose finden Sie unter: https://www.akdae.de/Arzneimitteltherapie/AVP/vorab/20200213-Osteoporose.pdf
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 15. April 2020 vorab online veröffentlicht.