Naldemedin (Rizmoic®) ▼
Zugelassene Indikation und Wirkmechanismus
Rizmoic® (Naldemedin) ist zur Behandlung von opioidinduzierter Obstipation (OIC, opioid-induced constipation) bei Erwachsenen zugelassen, die früher bereits mit einem Abführmittel behandelt wurden. Naldemedin ist ein Naltrexon-Derivat, das chemisch so verändert wurde, dass es die Blut-Hirn-Schranke nicht überqueren kann. Zudem ist Naldemedin ein Substrat des P-Glykoprotein (P-gp)-Effluxtransporters, der möglicherweise ebenfalls dazu beiträgt, dass Naldemedin nicht in das ZNS eindringt. Naldemedin antagonisiert die Opioid-Bindung an den µ-, δ- und κ-Opioidrezeptoren im Gastrointestinaltrakt und vermindert dadurch die obstipierenden Wirkungen von Opioiden ohne die ZNS-vermittelten Opioideffekte aufzuheben. Der pharmazeutische Unternehmer gab bei der Zulassung an, dass Naldemedin aufgrund seiner langsamen Assoziations- und Dissoziationskinetik am μ-Opioidrezeptor vermutlich als nicht kompetitiver Antagonist wirkt, sodass die antagonistische Wirkung auch bei höheren Opioidkonzentrationen aufrechterhalten bleibt. Diese Annahme basierte auf Ergebnissen präklinischer Studien. Der Ausschuss für Humanarzneimittel der EMA beschrieb im European Public Assessment Report Naldemedin aber eher als kompetitiver Antagonist, sodass ein sogenannter Ceiling-Effekt nicht ausgeschlossen werden kann.
Markteinführung
Rizmoic® (Naldemedin) ist seit dem 15. Mai 2020 in dieser Indikation auf dem deutschen Markt.
Bewertung
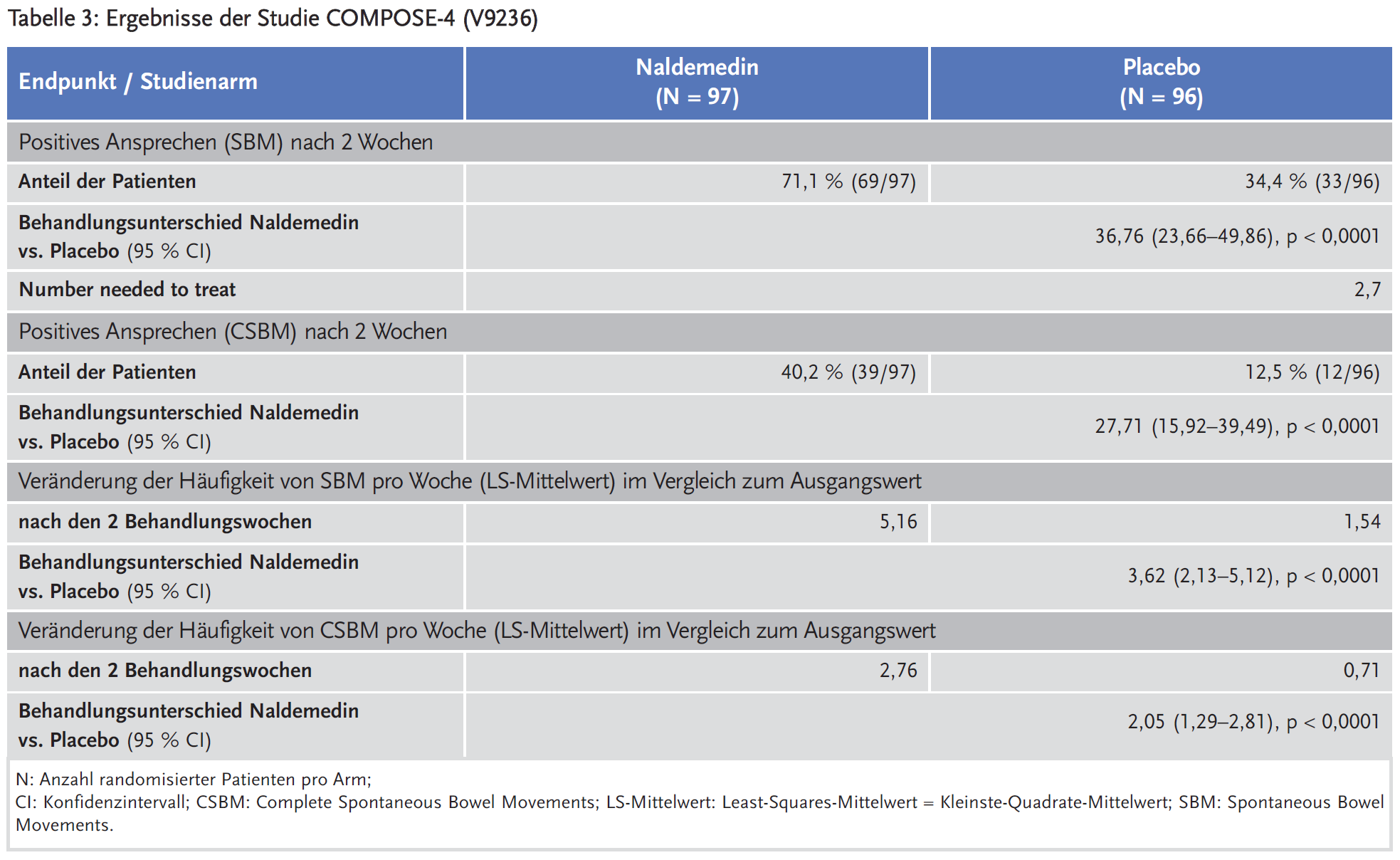
Naldemedin erhöhte bei Patienten mit nicht krebsbedingten Schmerzen und opioidinduzierter Obstipation die Häufigkeit spontaner Stuhlgänge (SBM) um 1,4 pro Woche und die Häufigkeit vollständiger spontaner Darmentleerungen (CSBM) um 1,1 pro Woche im Vergleich zu Placebo. Zudem wurde mit Naldemedin eine 75-prozentige Steigerung der Häufigkeit von SBM ohne Pressen pro Woche im Vergleich zu Placebo erreicht. Der Effekt von Naldemedin war bei Patienten mit Opioidbehandlung aufgrund von krebsbedingten chronischen Schmerzen stärker ausgeprägt. 71 % dieser Patienten erreichten bereits nach zwei Wochen ein positives Ansprechen mit ≥ 3 SBM pro Woche und einem Anstieg gegenüber dem Ausgangswert von mindestens einem SBM pro Woche. Die Häufigkeit der SBM und CSBM wurde statistisch signifikant um 3,6 bzw. 2,1 pro Woche im Vergleich zu Placebo erhöht.
Weder die von den Patienten berichtete Symptomatik (PAC-SYM) noch die Lebensqualität (PAC-QOL) wurde durch Naldemedin im Vergleich zu Placebo statistisch signifikant bzw. klinisch relevant verbessert. Ob Naldemedin eine bessere Wirksamkeit und Sicherheit im Vergleich zu konventionellen Laxanzien (und ihren Kombinationen), anderen peripher wirkenden μ-Opioidrezeptor-Antagonisten (PAMORA, Naloxegol und Methylnaltrexon) oder andere Arzneimittel gegen Obstipation (Linaclotid, Prucaloprid) aufweist, ist nicht zu beurteilen, weil die Studien nur im Vergleich zu Placebo durchgeführt wurden.
Naldemedin zeigte in den Studien eine gute Verträglichkeit mit Nebenwirkungen wie z. B. Abdominalschmerzen, Diarrhoe, Übelkeit und Erbrechen sowie Opioidentzugssyndrom. Bei den Patienten, die Methadon erhielten (4,2 % im Naldemedin-Arm; 4, 7 % im Placebo-Arm), traten im Vergleich zu den Untergruppen mit anderen Opioiden deutlich häufiger Nebenwirkungen wie Abdominalschmerzen, Diarrhoe, Übelkeit und Erbrechen auf. Die Sicherheit von Naldemedin ist bei gleichzeitiger Behandlung mit Methadon daher möglicherweise beeinträchtigt. Zudem ist die Langzeitsicherheit von Naldemedin derzeit nicht abschließend zu bewerten, da nur eine Studie über 52 Wochen durchgeführt wurde. Die Sicherheit von Naldemedin bei Patienten mit schweren Leberfunktionsstörungen, Kindern, Patienten im Alter ≥ 75 Jahre und solchen mit kardiovaskulären Risikofaktoren ist derzeit nicht beurteilbar, weil keine Daten dazu vorliegen.
Es ist unklar, ob Naldemedin bei Patienten wirkt, die sehr hohe Dosen von Opioiden (> 400 mg Morphinäquivalenzdosen) erhalten. Bei Patienten, die mit dem μ-Opioidrezeptor-Partialagonisten Buprenorphin behandelt werden, ist anzunehmen, dass die Wirksamkeit von Naldemedin verringert ist. Zudem ist unklar, ob die Wirksamkeit von Naldemedin über einen längeren Zeitraum aufrechterhalten wird; derzeit liegen Daten zu maximal 52 Wochen vor.
Der Stellenwert von Naldemedin ist derzeit nicht beurteilbar, sodass Naldemedin nur bei Patienten mit opioidinduzierter Obstipation einzusetzen ist, die bereits mit Laxanzien behandelt wurden und diese nicht ausreichend wirken oder nicht vertragen werden.
Wirksamkeit in den Zulassungsstudien
Die Zulassung von Naldemedin basierte auf den vier COMPOSE-Studien. Zwei multizentrische, doppelblinde, randomisierte, placebokontrollierte Studien mit Parallelgruppendesign – COMPOSE-1 und COMPOSE-2 – bestanden aus einer zwei- bis vierwöchigen Screening-Phase, einer zwölfwöchigen Behandlungsphase und einer vierwöchigen Nachbeobachtung. Eingeschlossen wurden erwachsene Patienten mit nicht krebsbedingten chronischen Schmerzen unter Opioidtherapie, die an opioidinduzierter Verstopfung litten. Ein Beigebrauch von Laxanzien war nicht erlaubt. Eine Woche mit positivem Ansprechen war definiert durch mindestens drei spontane Stuhlgänge (Spontaneous Bowel Movements, SBM) und einen Anstieg gegenüber dem Ausgangswert von mindestens einem SBM pro Woche. Der primäre Endpunkt der Studien war operationalisiert als mindestens neun Wochen mit positivem Ansprechen in der zwölfwöchigen Behandlungsperiode und drei Wochen mit positivem Ansprechen in den letzten vier Wochen der zwölfwöchigen Behandlungsperiode. Als sekundäre Endpunkte wurde u. a. die Veränderung der Häufigkeit der SBM pro Woche, der Anzahl vollständiger spontaner Darmentleerungen (Complete Spontaneous Bowel Movements, CSBM) pro Woche und der SBM ohne Pressen erhoben (Tabelle 1). Das Durchschnittsalter der Patienten in den Studien betrug 53,2 Jahre; 14,8 % waren 65 Jahre oder älter; 62,0 % waren Frauen und 80,2 % waren weiß. Die drei häufigsten Schmerzarten in COMPOSE-1 waren Rückenschmerzen (62,0 %), Nackenschmerzen (8,3 %) und Osteoarthrose (5,3 %). In COMPOSE-2 waren es Rückenschmerzen (53,6 %); Schmerz (10,2 %) und Arthralgie (7,8 %). Die durchschnittliche tägliche Morphinäquivalenzdosis des jeweiligen Opioids betrug zu Studienbeginn 132,42 mg in COMPOSE-1 und 120,93 mg in COMPOSE-2.
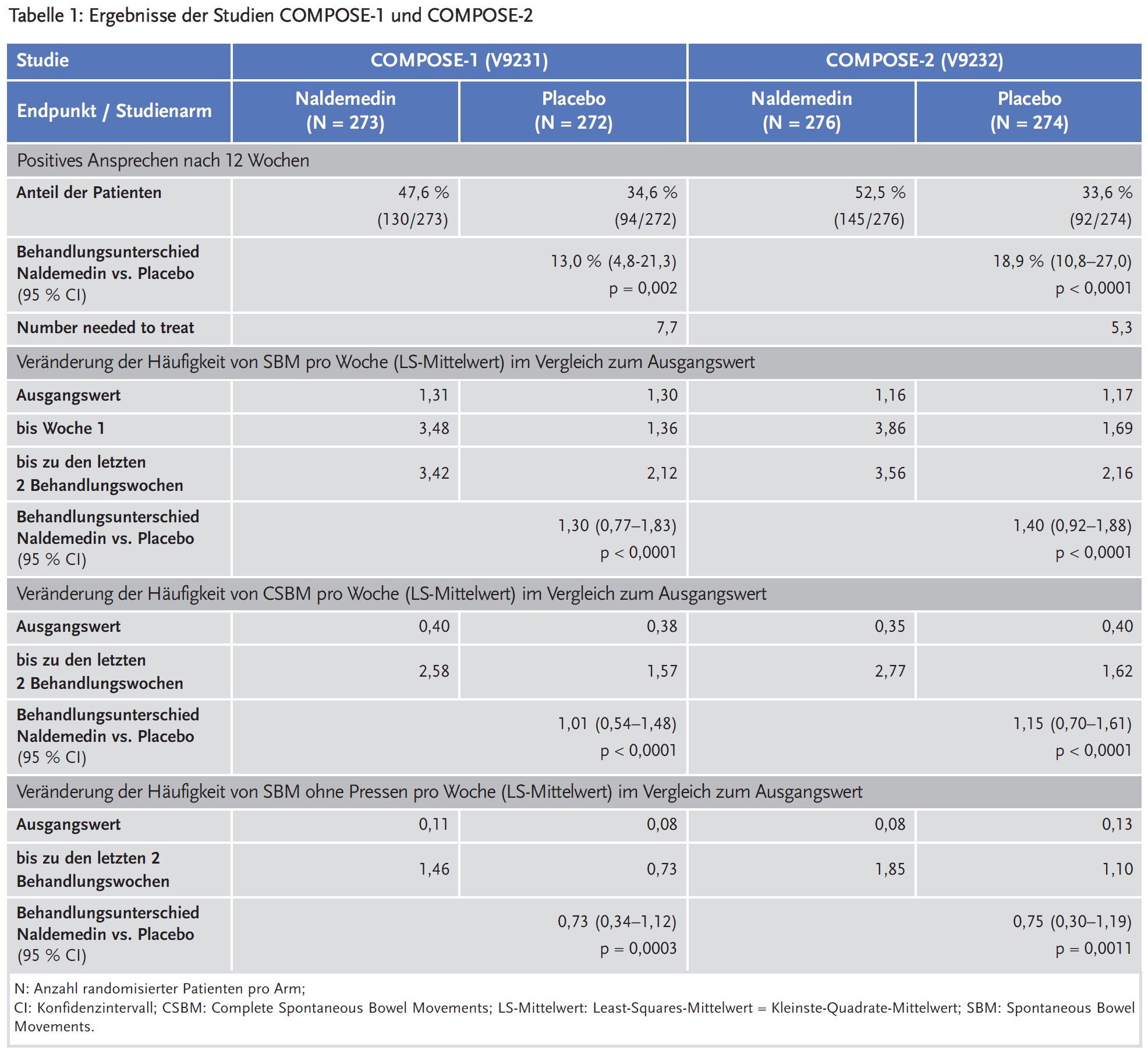
Die Wirksamkeit von Naldemedin wurde gepooled für beide Studien auch in zum Teil post hoc definierten Subgruppen untersucht, z. B. bei Patienten, die nicht auf Laxanzien ansprachen (Laxative Inadequate Responders(LIR)-Subgruppe bzw. Nicht-LIR-Subgruppe. Die Einstufung als LIR erfolgte auf der Grundlage von Informationen über die Begleitmedikationen vor Eintritt in die Studie und die Behandlung mit Laxanzien (Naldemedin: n = 317; Placebo: n = 312). Patienten, die innerhalb von 30 Tagen vor Screening keine Laxanzien erhielten und danach nur mit einem Notfall-Laxativum behandelt wurden, wurden als Nicht-LIR betrachtet (Naldemedin: n = 223; Placebo: n = 228). Der Anteil der SBM-Responder unter Naldemedin (46,4 %) war in der LIR-Subgruppe nummerisch niedriger als in der Nicht-LIR-Subgruppe (54,3 %), der Behandlungsunterschied zu Placebo war aber in beiden Gruppen vergleichbar (16,2 % vs. 15,6 %). Bezüglich des Anteils der CSBM-Responder zeigte sich eine deutlichere Differenz im Behandlungsunterschied zu Placebo: 10,5 % in der LIR-Subgruppe vs. 15,1 % in der Nicht-LIR-Subgruppe).
Durchgeführt wurde eine weitere randomisierte, doppelblinde, placebokontrollierte Langzeitstudie über 52 Wochen (COMPOSE-3), in der Naldemedin mit Laxanzien in stabiler Dosierung (50,6 %) oder ohne Laxanzien (30,0 %) bei Patienten mit chronischen, nicht krebsbedingten Schmerzen und opioidinduzierter Verstopfung getestet wurde. Patienten, die zum Zeitpunkt der Screening-Untersuchung ein stabiles Behandlungsschema mit Laxanzien anwendeten (50,6 % im Naldemedin-Arm; 54,2 % im Placebo-Arm), war es erlaubt, diese über die gesamte Studiendauer unverändert weiter anzuwenden. Die drei häufigsten Schmerzarten waren Rückenschmerzen (58,0 %), Osteoarthrose (9,5 %) und Nackenschmerzen (8,1 %). Die durchschnittliche tägliche Morphinäquivalenzdosis des jeweiligen Opioids betrug zu Studienbeginn 122,06 mg. Die mediane Anzahl von SBM pro Woche bei Studienbeginn betrug 1,60. Der primäre Endpunkt war die Sicherheit und Verträglichkeit von Naldemedin bei längerem Gebrauch. Als sekundärer Endpunkt wurde u. a. die Veränderung der Häufigkeit der Stuhlgänge pro Woche in der Woche 12, 24, 36 und 52 im Vergleich zum Ausgangswert evaluiert. Zudem wurden die Lebensqualität (anhand des Patient Assessment of Constipation Quality of Life, PAC-QOL) und der Patient Assessment of Constipation-Symptoms(PAC-SYM)-Score erhoben (Tabelle 2).1
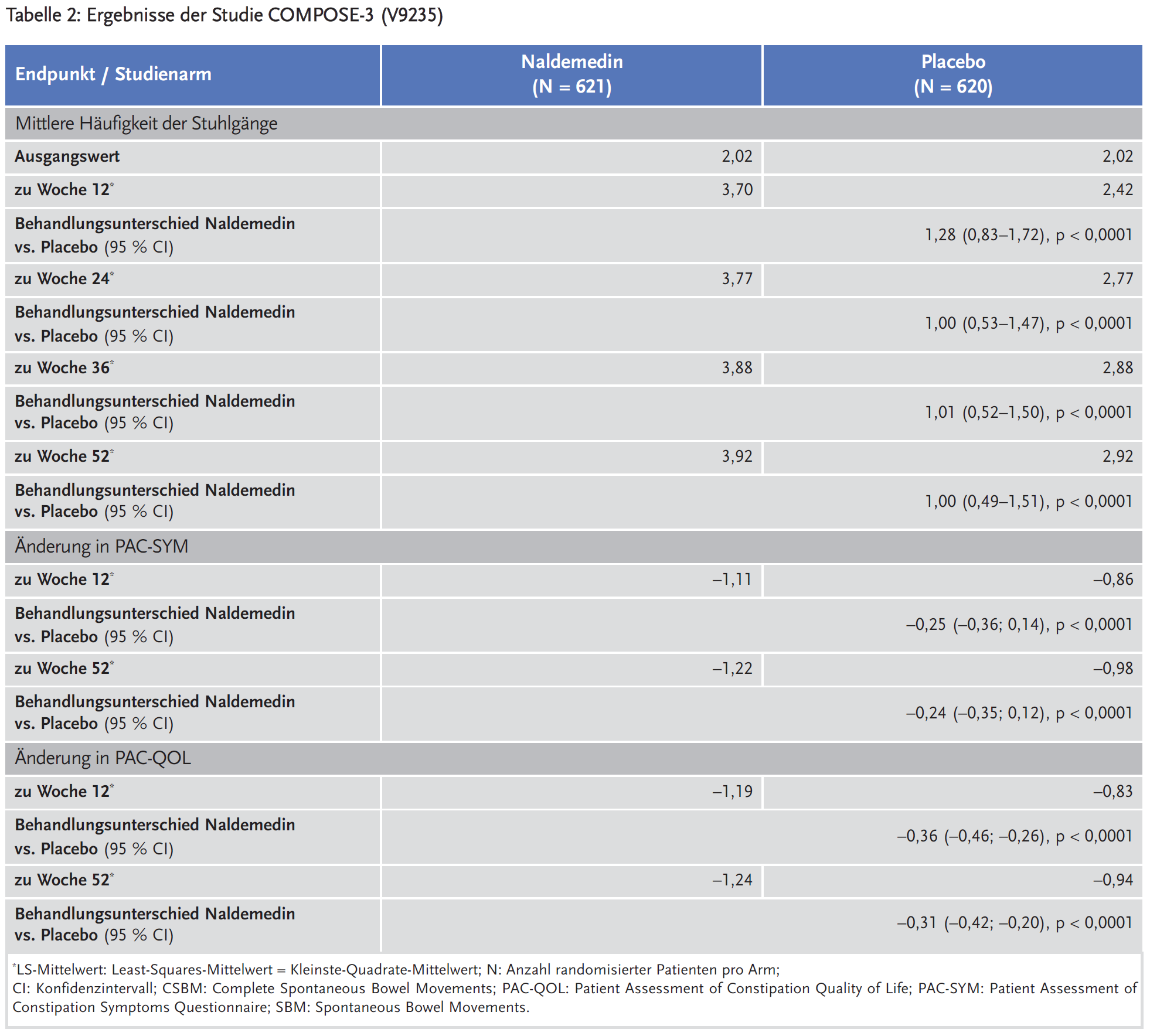
Auch in der Studie V9235 wurden Subgruppenanalysen durchgeführt u. a. bezüglich der Veränderung der Stuhlgangshäufigkeit in Woche 52 gegenüber dem Ausgangswert. Der Behandlungsunterschied Naldemedin vs. Placebo fiel sowohl in der LIR-Subgruppe (3,10 vs. 1,90; p = 0,0210) als auch in der Nicht-LIR-Subgruppe (4,26 vs. 3,39; p = 0,1349) zugunsten von Naldemedin aus, war aber in der Nicht-LIR-Subgruppe nicht statistisch signifikant.
Naldemedin wurde in einer doppelblinden, randomisierten, placebokontrollierten Studie (COMPOSE-4) an Patienten mit einer Krebserkrankung und opioidinduzierter Obstipation untersucht. Die Patienten mussten vor der Screening-Untersuchung ≥ 14 Tage mit Opioiden in einer stabilen Dosis behandelt worden sein. Die Studien bestanden aus einer zweiwöchigen Screeningphase, einer zweiwöchigen Behandlungsphase und einer vierwöchigen Nachbeobachtungsphase. Patienten, die beim Screening Laxanzien erhielten, mussten diese Therapie bis zum Ende des Behandlungszeitraums in einer stabilen Dosis fortsetzen. Zudem durften alle Patienten nach Bedarf Notfall-Laxanzien anwenden. Primärer Endpunkt war auch in dieser Studie der Anteil der Patienten mit positivem Ansprechen, definiert durch ≥ 3 SBM pro Woche und einen Anstieg gegenüber dem Ausgangswert von mindestens einem SBM pro Woche während der zweiwöchigen Behandlungsphase. Als sekundäre Punkte wurde u. a. erhoben die Ansprechrate bezüglich CSBM (≥ 3 CSBM pro Woche und Anstieg gegenüber dem Ausgangswert ≥ 1 CSBM pro Woche während der zweiwöchigen Behandlungsphase) sowie Veränderung der Häufigkeit der SBM pro Woche und der CSBM pro Woche (Tabelle 3). Erhoben wurden auch die Lebensqualität (PAC-QOL) und Symptomatik (PAC-SYM-Score), allerdings wurden einzelne Ergebnisse dazu nicht im EPAR berichtet. Es wird nur darauf hingewiesen, dass sich keine Unterschiede diesbezüglich zwischen den Behandlungsarmen gezeigt haben.
Ausgewählte Nebenwirkungen
Die häufigsten Nebenwirkungen sind Abdominalschmerzen, Diarrhoe, Übelkeit und Erbrechen. Gelegentlich tritt ein Opioidentzugssyndrom auf.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Naldemedin ist kontraindiziert bei Patienten mit bekannter oder vermuteter gastrointestinaler Obstruktion oder Perforation oder bei Patienten mit erhöhtem Risiko für eine wiederkehrende Obstruktion aufgrund der Gefahr einer gastrointestinalen Perforation.
- Naldemedin soll mit Vorsicht angewendet werden bei Patienten mit Erkrankungen, die zu einer strukturellen Schädigung der Wand des GI-Trakts führen könnten (z. B. peptische Ulkuskrankheit, Ogilvie-Syndrom (Pseudoobstruktion des Dickdarms), maligne Erkrankungen des GI-Trakts, Morbus Crohn).
- Unter Naldemedin kann eine Opioidentzugssyndrom auftreten mit drei oder mehr der Symptome: Dysphorie, Übelkeit oder Erbrechen, Muskelschmerzen, Tränensekretion oder Rhinorrhoe, Pupillenerweiterung oder Piloerektion oder Schwitzen, Diarrhoe, Gähnen, Fieber oder Schlaflosigkeit. In solchen Fällen ist die Behandlung mit Naldemedin abzusetzen.
- Patienten mit Störungen der Blut-Hirn-Schranke aufgrund von z. B. primären malignen Hirntumoren, ZNS-Metastasen oder anderen entzündlichen Erkrankungen, aktiver multipler Sklerose oder fortgeschrittener Alzheimer-Krankheit können ein erhöhtes Risiko für Opioidentzug oder eine verminderte analgetische Wirkung haben. Bei diesen Patienten ist eine engmaschige Überwachung auf Opioidentzugssymptome durchzuführen.
- Naldemedin wurde bei Patienten mit schwerer Beeinträchtigung der Leberfunktion nicht untersucht. Die Anwendung von Naldemedin bei diesen Patienten wird nicht empfohlen.
- Es liegen nur begrenzte Erfahrungen bei Patienten vor, die mit einem oder mehreren opioidhaltigen Schmerzmitteln behandelt werden, deren tägliche Dosierung mehr als 400 mg Morphin entspricht. Bei höheren Dosen ist die Wirkung derzeit unklar.
- Es liegen keine Erfahrungen bei Patienten vor, die wegen einer durch µ-Opioidrezeptor-Partialagonisten (z. B. Buprenorphin) verursachten Verstopfung behandelt werden. Es ist möglich, dass Naldemedin in diesen Fällen nicht wirksam ist.
- Patienten mit innerhalb von drei Monaten vor dem Screening aufgetretenem Myokardinfarkt, Schlaganfall oder transitorischer ischämischer Attacke wurden aus den Studien ausgeschlossen und sollten während der Einnahme von Naldemedin klinisch überwacht werden.
- Naldemedin muss abgesetzt werden, wenn die Behandlung mit dem Opioid-Analgetikum beendet wird.
- Naldemedin wird einmal täglich zu einer Mahlzeit oder unabhängig von Mahlzeiten zu jeder beliebigen Tageszeit eingenommen; empfehlenswert ist die Einnahme zur gleichen Uhrzeit.
- Naldemedin kann mit Laxanzien kombiniert werden.
- Naldemedin wird durch CYP3A verstoffwechselt, unter Beteiligung des Enzyms UGT1A3, und ist Substrat des P-Glykoproteins (P-gp). Die gleichzeitige Anwendung mit starken CYP3A-Inhibitoren (wie z. B. Grapefruitsaft, Johanniskraut, Rifampicin, Carbamazepin, Phenobarbital und Phenytoin) sollte vermieden werden, weil sie zu einem Anstieg der Naldemedin-Exposition führt und das Risiko für Nebenwirkungen erhöhen kann.

Weiterführende Informationen
Das IQWiG wurde am 15.05.2020 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Rizmoic®, erschienen am 1. März 2019. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Fußnoten
1Patient Assessment of Constipation Quality of Life (PAC-QOL): ein krankheitsspezifischer Fragebogen für Patienten mit Obstipation, der aus 28 Fragen besteht, die auf 5-Stufen-Skalen mit Punktzahlen bewertet werden. Erfasst werden 12 Symptome, die in vier Subskalen unterteilt sind: physisches Unbehagen (4 Fragen), psychosoziales Unbehagen (8 Fragen), Sorgen (11 Fragen) und Zufriedenheit (5 Fragen). Die Gesamtpunktzahl beträgt 0–112 Punkte. Je höher die erreichte Punktzahl ist, desto schlechter ist die Lebensqualität.
Patient Assessment of Constipation-Symptoms (PAC-SYM): 12-Punkte-Fragebogen, der durch die psychometrische Beurteilung von Erwachsenen mit Obstipation als Instrument zur Beurteilung des Schweregrades der von Patienten berichteten Symptome chronischer Obstipation entwickelt wurde. Der Fragebogen ist in drei Symptom-Subskalen unterteilt: abdominal (4 Fragen), rektal (3 Fragen) und Stuhl (5 Fragen). Die Fragen werden auf 5-Stufen-Skalen mit Punktzahlen von 0 bis 4 bewertet: 0 = Symptom fehlt; 1 = leicht; 2 = mäßig; 3 = schwer und 4 = sehr schwer. Eine mittlere Gesamtpunktzahl von 0–4 ergibt sich durch Division der Gesamtpunktzahl durch die Anzahl der beantworteten Fragen; je niedriger die Gesamtpunktzahl, desto geringer die Symptombelastung.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 22. Juni 2020 vorab online veröffentlicht.