Remdesivir (Veklury®) ▼
Zugelassene Indikation und Wirkmechanismus
Veklury® (Remdesivir) ist unter erheblichen Auflagen zugelassen zur Behandlung von COVID-19 bei Erwachsenen und Jugendlichen (im Alter von mindestens 12 Jahren und mit einem Körpergewicht von mindestens 40 kg) mit einer Pneumonie, die eine zusätzliche Sauerstoffzufuhr erfordert. Remdesivir ist ein Adenosin-Nukleotid-Prodrug, das in Wirtszellen zum wirksamen Nukleosid-Triphosphat-Metaboliten umgewandelt wird. Remdesivir-Triphosphat wirkt als ein Analogon von Adenosin-Triphosphat (ATP) und konkurriert mit dem natürlichen Substrat ATP um die Integration in entstehende RNA-Ketten durch die SARS-CoV-2-RNA-abhängige RNA-Polymerase. Dies führt zu einer verzögerten Kettenterminierung während der Replikation der viralen RNA und damit zur Inhibition der Virusreplikation.
Markteinführung
Veklury® (Remdesivir) ist noch nicht auf dem deutschen Arzneimittelmarkt verfügbar.
Bewertung
Veklury® (Remdesivir) hat in einer noch laufenden doppelblinden, randomisierten, kontrollierten Studie die Zeit bis zur Genesung (operationalisiert als Zeit bis zur Entlassung aus stationärer Behandlung) im Vergleich zu Placebo bei Patienten mit schwerem COVID-19 signifikant verkürzt. Bezüglich der Mortalität nach 14 Tagen zeigte sich bisher kein statistisch signifikanter Unterschied zu Placebo. Andere Studien – z. T. offen oder nicht ausreichend gepowered – konnten diesen positiven Effekt bis jetzt nicht bestätigen. Ergebnisse aus den anderen großen Studien mit adaptivem Design wie Solidarity und DisCoVeRy sind noch nicht veröffentlicht worden.
Remdesivir erscheint bisher gut verträglich, die verfügbaren, vorläufigen Daten zur Sicherheit sind aber nicht umfassend. Überempfindlichkeitsreaktionen und Erhöhung der Transaminasen treten häufig auf. Die Langzeitsicherheit bzw. Spätfolgen von Remdesivir können nicht abschließend beurteilt werden.
Die COVID-19-Pandemie stellt unbestreitbar ein bedeutendes Risiko für die öffentliche Gesundheit dar. Da bislang kein anderes Arzneimittel mit einer nachgewiesenen Wirksamkeit gegen COVID-19 in der EU zugelassen ist, besteht ein erheblicher ungedeckter medizinischer Bedarf. Der Einsatz von Remdesivir bei Patienten mit Pneumonie und zusätzlichem Sauerstoffbedarf, aber nicht bei Patienten mit NIV/„High-Flow“-Sauerstofftherapie oder invasiver Beatmung/ECMO, erscheint aufgrund der vorliegenden Daten derzeit gerechtfertigt. Dabei ist allerdings davon auszugehen, dass Remdesivir wahrscheinlich hauptsächlich bei jüngeren, nicht schwer erkrankten Patienten mit COVID-19, nicht aber bei schwer erkrankten Patienten auf Intensivstation zu einen Behandlungsvorteil im Sinne einer Verkürzung der stationären Aufenthaltsdauer führt. Ob die Behandlung mit Remdesivir einen Vorteil bezüglich der Mortalität für eine dieser Patientengruppen erbringt, ist derzeit unklar.
Wirksamkeit in den Zulassungsstudien
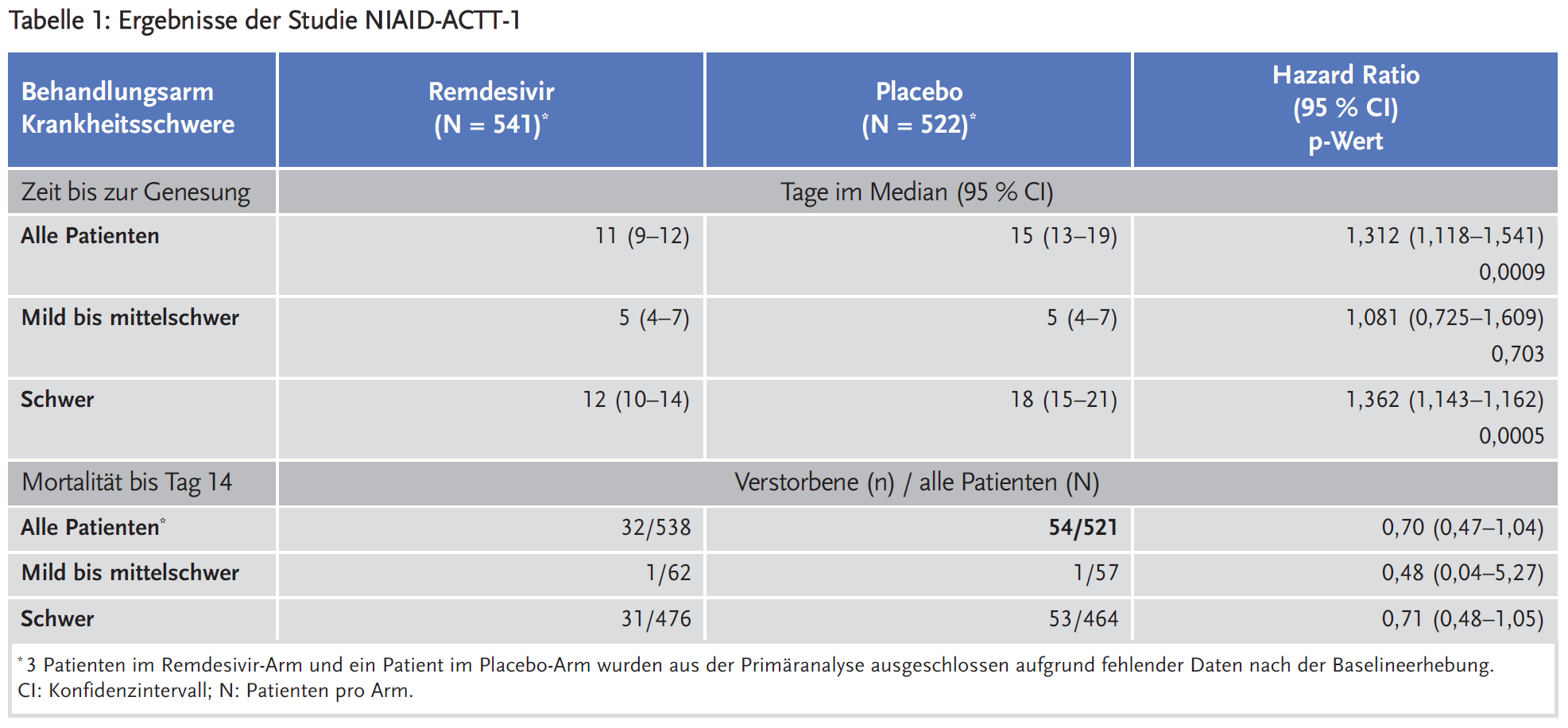
Für die Zulassung wurde die multizentrische, doppelblinde randomisierte kontrollierte Studie NIAID-ACTT-1 eingereicht. Eingeschlossen wurden Patienten mit nachgewiesener SARS-CoV-2-Infektion, die entweder Lungeninfiltrate (durch Bildgebung bestätigt) oder eine Sauerstoffsättigung (SpO2) von ≤ 94 % aufwiesen oder Sauerstoffgabe oder maschinelle Beatmung erforderten. Die Patienten wurden stratifiziert nach Schweregrad der Erkrankung: schwer: maschinelle Beatmung oder Sauerstoffgabe oder SpO2 ≤ 94 % oder Tachypnoe (Atemfrequenz ≥ 24/min); mild bis moderat: SpO2 > 94 % und Atemfrequenz 24/min ohne Sauerstoffgabe. Remdesivir erhielten 531 Patienten für maximal zehn Tage, 518 wurden mit Placebo behandelt. Das mittlere Alter betrug 58,9 Jahre; etwa zwei Drittel der Patienten waren männlich. Die Symptome bestanden seit im Median neun Tagen. Eine schwere Vorerkrankung wiesen 88,7 % der Patienten auf. Bluthochdruck hatten 49,6 % der Patienten, Asthma 11,4 %, Diabetes mellitus Typ 2 29,7 % und Adipositas 37 %. 11,9 % der Patienten benötigten keine Sauerstoffgabe, 39,6 % erhielten Sauerstoff, 18,5 % benötigten nicht invasive Beatmung (non-invasive ventilation, NIV) oder „High-Flow“-Sauerstofftherapie und bei 25,6 % war eine extrakorporale Membranoxygenierung (extracorporeal membrane oxygenation, ECMO) erforderlich.
Primärer Endpunkt der Studie war die Zeit bis zur Genesung; eine Genesung bestand laut Operationalisierung, wenn die Patienten entweder keinen Sauerstoff mehr benötigten oder nicht mehr hospitalisiert waren. Als sekundäre Endpunkte wurden die Mortalität am Tag 14 und 29 sowie der klinische Status am Tag 15 (tot / hospitalisiert (+ Beatmung) / nicht hospitalisiert) erhoben (Tabelle 1).
Für Remdesivir wurde eine sogenannte bedingte Zulassung erteilt („conditional marketing authorisation“, CMA). Für eine CMA sind weniger umfangreiche Daten als für eine reguläre Zulassung erforderlich. Diese Form der Zulassung kommt in bestimmten Situationen in Betracht, wenn das Arzneimittel zur Behandlung einer seltenen und/oder lebensbedrohlichen Krankheit bestimmt ist und das Nutzen-Risiko-Verhältnis als positiv eingestuft wurde. Die Anforderungen an eine bedingte Zulassung von Remdesivir sind erfüllt, da bei COVID-19 ein ungedeckter medizinischer Bedarf besteht („unmet medical need“) und es sich um ein Arzneimittel handelt, zu dem noch unvollständige klinische Daten vorliegen und das in Krisensituationen gegen eine Bedrohung der öffentlichen Gesundheit eingesetzt werden soll (1). Eine CMA wird jährlich neu bewertet. Um sie in eine reguläre Zulassung zu überführen, müssen weitere Daten vorgelegt werden. Der pharmazeutische Unternehmer Gilead wurde verpflichtet, finale Ergebnisse zur Mortalität in der NIAID-ACTT-1-Studie bis August 2020 und zu allen klinischen Studien mit Remdesivir bis Dezember 2020 vorzulegen (1).
Kritik an der Zulassungsstudie
Die Daten der NIAID-ACTT-1-Studie (2) wurden bisher nur als Preliminary Report publiziert. Sie beruhen auf einer vorläufigen Datenauswertung, weitere Analysen mit weitergehender Bewertung bezüglich patientenrelevanter Endpunkte wie Mortalität sowie Daten aus anderen großen laufenden Studien stehen noch aus.
- Das Durchschnittsalter der Studienpopulation war mit 59 Jahren deutlich jünger als das Alter der stationären SARS-CoV-2-Patienten in deutschen Kliniken. Zwar liegt laut Robert Koch-Institut der Altersmedian bei den COVID-19-Erkrankten bei 49 Jahren, allerdings besteht eine deutliche Korrelation zwischen dem Patientenalter und schweren Verläufen (Intensivbehandlung mit Beatmungspflichtigkeit) sowie COVID-19-bedingter Mortalität. In Deutschland ist die Mehrzahl der Patienten mit COVID-19 auf Intensivstation sogar über 80 Jahre alt (3).
- Die in der Studie vorgenommene dichotome Einteilung nach Schweregrad der Erkrankung in mild bis moderat (SpO2 > 94 % und Atemfrequenz < 24/min ohne Sauerstoffgabe) versus schwer (maschinelle Beatmung oder Sauerstoffgabe oder SpO2 ≤ 94 % oder Tachypnoe (Atemfrequenz ≥ 24/min)) ist eine Unterteilung der Patienten in Gruppen, die sich so im klinischen Alltag nicht wiederfindet. Eine Einteilung in mild (kein zusätzlicher Sauerstoffbedarf; ambulante Behandlung), moderat (zusätzlicher Sauerstoffbedarf, Hospitalisierung auf Normalstation bzw. unter besonderer Überwachung), schwer (NIV/„High-Flow“-Sauerstofftherapie, Hospitalisierung auf Intensivstation) und kritisch (Intubation/ECMO, Hospitalisierung auf Intensivstation) wäre geeigneter gewesen, um die Wirksamkeit für die einzelnen Patientengruppen im Hinblick auf die klinische Realität zu bewerten. Dies würde Ärzten ermöglichen, die Ergebnisse, die zur Zulassung geführt haben, rational für ihre Patienten umzusetzen. Eine solche Schweregradeinteilung korreliert mit dem Ausmaß der Infektion bzw. der immunologisch vermittelten Inflammation und den Erkrankungsphasen. Patienten mit apparativer Atmungsunterstützung befinden sich meist in der späten Erkrankungsphase mit hyperinflammatorischer Reaktion, in der eine antivirale Therapie in der Regel keinen Effekt mehr hat. Eine antivirale Therapie sollte dementsprechend eher in der frühen infektiösen Erkrankungsphase (mit pulmonaler Beteiligung, daher sauerstoffpflichtig) zum Einsatz kommen. Eine Zusammensetzung der beiden Gruppen – wie sie in der Studie konzipiert wurde – ist nicht zielführend und vermischt pathophysiologisch unterschiedlich ausgelöste Krankheitsverläufe.
- Die gewählte Definition für Genesung ist für einen primären Endpunkt zu ungenau, weil es letztlich nur Entlassung aus stationärer Behandlung bedeutet, unabhängig vom Zustand des Patienten. Die Zeit bis zum Erreichen klinischer Stabilität, für die es bei ambulant erworbener Pneumonie klare, evidenzbasierte Kriterien gibt, wäre daher als primärer Endpunkt zielführender gewesen. Tatsächlich ergibt sich aus einem Histogramm im Supplementary Appendix (4) zur Originalpublikation, dass der größte Teil der am Tag 15 als genesen eingestuften Patienten nach Entlassung noch in der Kategorie 2 verblieb (weiterhin Sauerstoffbedarf und/oder Einschränkung körperlicher Aktivitäten).
- Zudem war die Größe der einzelnen Patientengruppen in der Studie nicht ausreichend. Im Remdesivir-Arm erhielten insgesamt 222 Patienten Sauerstoffbehandlung, 98 NIV/„High-Flow“-Sauerstofftherapie und 125 eine invasive Beatmung/ECMO. Keine der Odds Ratios für die Patientengruppe mit invasiver Beatmung/ECMO, für NIV/„High-Flow“-Sauerstofftherapie oder Sauerstoffbehandlung war für sich statistisch signifikant. Wie viele von den Patienten in jeder Gruppe den Endpunkt – Verkürzung der Zeit bis zur Genesung – erreichten, ist nicht berichtet. Es kann daher davon ausgegangen werden, dass die Gruppengröße zu klein war, um daraus eine statistisch signifikante Aussage abzuleiten. Vielmehr wurde dieses Problem durch eine „Mischung“ der Patientengruppen in der Studie gelöst.
- Die Patienten in der Studie erhielten Remdesivir im Mittel neun Tage nach Symptombeginn. Es scheint, dass der Einsatz zu diesem späten Zeitpunkt generell weniger aussichtsreich ist. Hierfür sprechen sowohl der Wirkmechanismus von Remdesivir als auch die Erfahrungen mit den Neuraminidasehemmern bei Influenza.
- Bei detaillierter Betrachtung der Kaplan-Meier-Kurven zum primären Endpunkt – Zeit bis zur sogenannten Genesung – ist klar zu erkennen, dass ein Behandlungsvorteil eben nur für die Patientengruppe mit Sauerstoffbedarf, aber nicht für Patienten mit NIV/„High-Flow“-Sauerstofftherapie oder invasiver Beatmung/ECMO besteht. Die Zulassung wurde aber erteilt für alle Patienten mit einer COVID-19-Pneumonie, die eine zusätzliche Sauerstoffzufuhr erfordert, und impliziert daher, dass auch Patienten auf Intensivstationen davon profitieren. Dies entspricht nicht den vorläufigen Ergebnissen der Studie.
Ausgewählte Nebenwirkungen
Die häufigsten Nebenwirkungen von Remdesivir bei gesunden, freiwilligen Probanden waren erhöhte Transaminasen (14 %) und bei Patienten mit COVID-19 Übelkeit (4 %). Häufige Nebenwirkungen sind Kopfschmerzen und Hautauschlag. Aufgetreten sind auch Überempfindlichkeitsreaktionen und infusionsbedingte Reaktionen.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Bei Patienten über 65 Jahren ist keine Dosisanpassung von Remdesivir erforderlich.
- Remdesivir sollte bei Patienten mit einer eGFR < 30 ml/min nicht angewendet werden.
- Es wurden Überempfindlichkeitsreaktionen einschließlich infusionsbedingter und anaphylaktischer Reaktionen während und nach der Anwendung von Remdesivir beobachtet. Langsamere Infusionsraten mit einer maximalen Infusionszeit von bis zu 120 Minuten können erwogen werden, um diesen Reaktionen potenziell vorzubeugen.
- Die Behandlung mit Remdesivir sollte nicht bei Patienten mit einem Alanin-Aminotransferase (ALT)-Wert ≥ dem 5-Fachen der normalen Obergrenze zu Therapiebeginn begonnen werden.
- Die Therapie mit Remdesivir sollte bei Patienten abgebrochen werden, wenn der ALT-Spiegel ≥ dem 5-Fachen der normalen Obergrenze ansteigt oder der ALT-Anstieg mit Anzeichen einer Leberentzündung oder ansteigendem konjugiertem Bilirubin, ansteigender alkalischer Phosphatase oder ansteigender INR einhergeht.
- Die gleichzeitige Anwendung von Remdesivir und Chloroquin oder Hydroxychloroquin wird aufgrund von In-vitro-Daten, die eine antagonistische Wirkung von Chloroquin auf die intrazelluläre Stoffwechselaktivierung und antivirale Aktivität von Remdesivir nachweisen, nicht empfohlen.
- Remdesivir ist in-vitro ein Substrat für CYP2C8, CYP2D6 und CYP3A4 und den organischen Anion-Transporter OATP1B1 sowie den P-gp-Transporter sowie ein Hemmer von CYP3A4, OATP1B1 und OATP1B3. Die klinische Relevanz dieser In-vitro-Arzneimittelwechselwirkungen ist derzeit unklar.
Weiterführende Informationen
Das IQWiG wurde am 15.05.2020 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Veklury®, erschienen am 6. Juli 2020. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Literatur
- Remdesivir als erstes Medikament von der Europäischen Kommission zur Behandlung von Patienten mit COVID-19 zugelassen: Mehr als ein Hoffnungsträger? Arzneimittelbrief 2020; 54: 56.
- Beigel JH, Tomashek KM, Dodd LE et al.: Remdesivir for the treatment of Covid-19 – preliminary report. N Engl J Med 2020; Epub ahead of print: NEJM oa2007764.
- Robert Koch-Institut (RKI) (Hrsg.): Epidemiologisches Bulletin. Krankheitsschwere von COVID-19, Nowcasting: Erkrankungsfälle und Reproduktionszahl. Epidemiol Bull 2020; 17.
- Beigel JH, Tomashek KM, Dodd LE et al.: Remdesivir for the treatment of Covid-19 - Preliminary Report (Supplementary appendix). N Engl J Med 2020; Epub ahead of print: NEJMoa2007764.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 6. August 2020 vorab online veröffentlicht.