Beschleunigte Zulassung neuer Arzneimittel: Königsweg oder Sackgasse?
In den letzten Jahren haben amerikanische und europäische Zulassungsbehörden mit verschiedenen Maßnahmen eine Beschleunigung der Zulassung von neuen Arzneimitteln angestrebt. Grundlage dafür ist die Annahme, dass ein schnellerer Zugang zu neuen Arzneimitteln Vorteile für Patienten bietet. Das mit diesen Maßnahmen verbundene Narrativ von „Neuheit“ und „Innovation“ erweckt die Erwartung, dass neue Produkte besser seien als bereits existierende Therapieoptionen. Davon abweichend haben Untersuchungen seit den 1970er Jahren gezeigt, dass nur ein kleiner Teil der neuen Arzneimittel zu wirklichen Fortschritten führt.
Vor diesem Hintergrund hat das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) die Ergebnisse der frühen Nutzenbewertung von Arzneimitteln nach dem Arzneimittelmarktneuordnungsgesetzt (AMNOG) (§ 35a SGB V) ausgewertet und in einer Publikation im Britisch Medical Journal beschrieben. Die Arbeit fasst die Ergebnisse der Jahre 2010 bis 2017 zusammen und stellt sie in den Kontext der aktuellen Diskussion um Arzneimittelentwicklung und -zulassung (1).
In den Jahren 2011 bis 2017 hat das IQWiG 216 neue Arzneimittel bewertet, darunter 152 neue Arzneimittel, deren Wirkstoffe erstmals zugelassen wurden, und 64 Arzneimittel, die eine Zulassung für eine zusätzliche Indikation erhielten. Nur für 54 der 216 bewerteten Arzneimittel (25 %) ergab die Bewertung des IQWiG einen beträchtlichen oder erheblichen Zusatznutzen in der zugelassenen Patientenpopulation. In 35 Fällen (16 %) wurde der Zusatznutzen als gering oder nicht quantifizierbar eingestuft. Für 125 neue Arzneimittel (58 %) konnte auf Basis der vorhandenen Evidenz kein Zusatznutzen bezüglich der Mortalität, Morbidität oder gesundheitsbezogenen Lebensqualität im Vergleich zu der vom Gemeinsamen Bundesauschuss (G-BA) festgelegten zweckmäßigen Vergleichstherapie festgestellt werden. Eine Aktualisierung der Analyse bis Ende 2019 zeigt ähnliche Ergebnisse (beträchtlicher oder erheblicher Zusatznutzen: 79/337 [23 %] der Bewertungen, Abbildung 1).
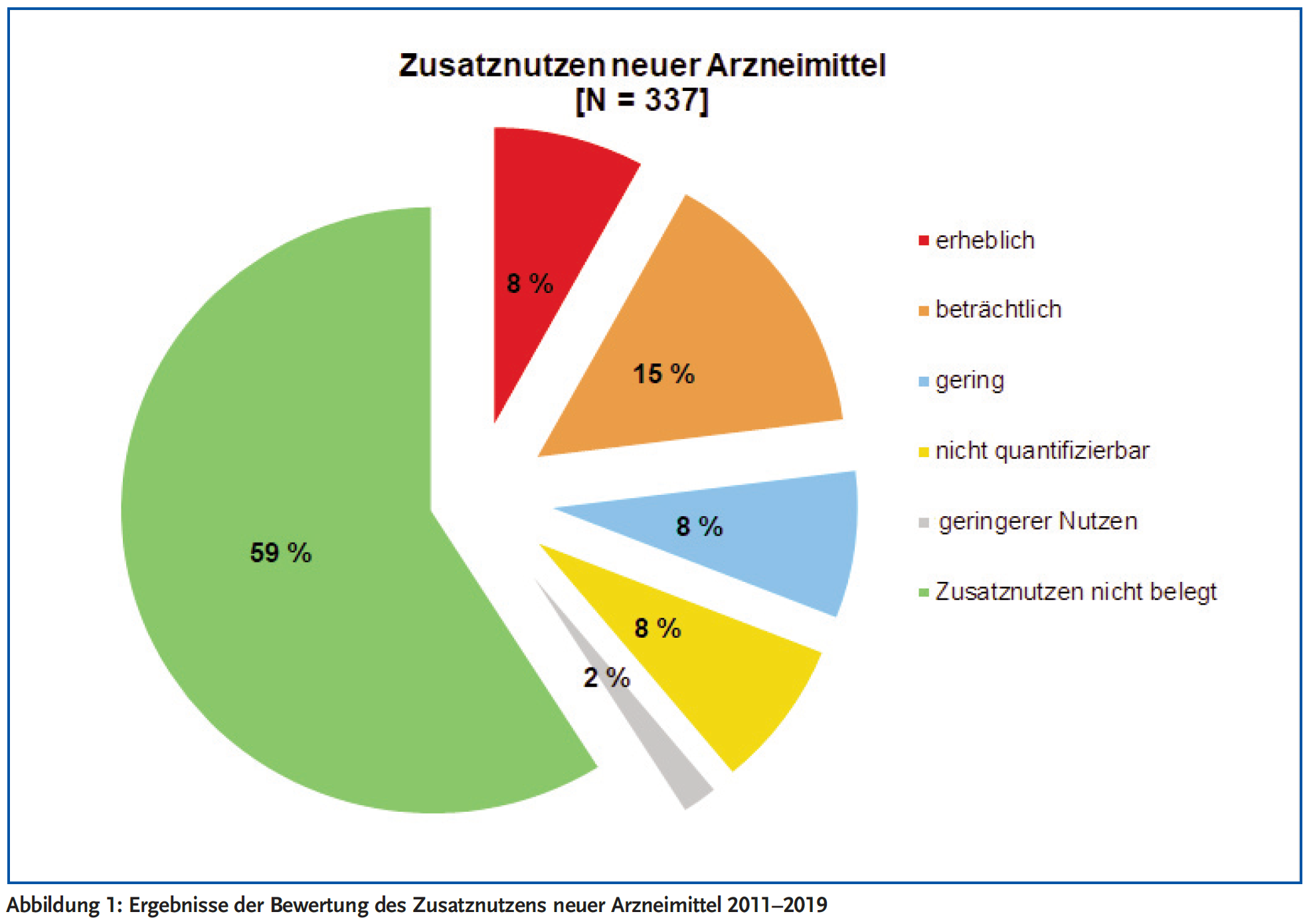
Die Ergebnisse unterscheiden sich zwischen den Indikationsgebieten. Während in der Onkologie auf Basis neuer Therapieprinzipien in 59 % (48/82) Bewertungen ein Zusatznutzen konstatiert wurde, sind die Ergebnisse in der Psychiatrie/Neurologie oder beim Diabetes mellitus (Nachweis eines Zusatznutzens für 6 % [1/18] und 17 % [4/24] der Bewertungen) besonders enttäuschend.
Was bedeuten diese Ergebnisse für neue Arzneimittel, die in Europa zugelassen werden? Ein geringerer Nutzen im Vergleich zum Therapiestandard wurde nur für 2 Arzneimittel (1 %) nachgewiesen – aber für 125 neue Arzneimittel können wir nicht beurteilen, ob sie einen geringeren, einen vergleichbaren oder einen höheren Nutzen für Patienten haben als die bereits verfügbaren Therapieoptionen. Dabei lagen für 64 neue Arzneimittel keine Studien für einen Vergleich mit der Standardtherapie vor. Für 42 gab es zwar entsprechende aktiv kontrollierte Studien, der Vergleich war aber nicht geeignet, die Frage nach dem Zusatznutzen zu beantworten, z. B. weil das Arzneimittel im Vergleichsarm in einer ungeeigneten Dosierung eingesetzt wurde. Für die verbleibenden 19 Fälle ohne Nachweis eines Zusatznutzens gab es geeignete Studien, diese zeigten aber keinen Unterschied zwischen dem neuen Arzneimittel und der Standardtherapie.
Evidenzgenerierung nach Zulassung: Illusion statt Lösung
Die Befürworter beschleunigter Zulassungen argumentieren, dass begrenzte Informationen zum Zeitpunkt des Markeintritts (und damit der Behandlung von Patienten) in Kauf genommen werden müssten, um einen schnellen Zugang zu innovativen Arzneimitteln zu ermöglichen. Dabei wird davon ausgegangen, dass die Erhebung von Daten nach der Zulassung den Nutzen für die Patienten zeigen wird.
Die Realität bestätigt diese Hoffnung nicht. Verschiedene systematische Untersuchungen der Evidenz, die nach Zulassung generiert wird, zeigen, dass die offenen Fragen in den wenigsten Fällen beantwortet werden. Auch die Erfahrungen innerhalb der Bewertungen nach dem AMNOG sind ernüchternd: Keine der zwischen 2011 und 2017 vom G-BA angeforderten Studien wurde durchgeführt.
Auswirkungen auf Patienten und Gesundheitssysteme
Patienten und ihre Ärzte benötigen vollständige und unabhängige Informationen darüber, was sie von neuen Arzneimitteln erwarten können. Dazu gehören auch Informationen über den Verlauf der Erkrankung unter alternativen Behandlungsoptionen, ggf. einschließlich der Option, keine Behandlung durchzuführen. Angesichts der bestehenden Wissenslücken kann diese Information nicht zur Verfügung gestellt werden. Dadurch wird es für Patienten in vielen Fällen unmöglich, Entscheidungen gemäß ihren Präferenzen zu treffen. Die Informationslücken schaden auch dem Gesundheitssystem, da sie Entscheidungen auf Systemebene und die Qualität der Versorgung beeinträchtigen.
Neue Ansätze für die Arzneimittelentwicklung
Da Arzneimittelentwicklung, -zulassung, -erstattung und -preisbildung umfangreich reguliert werden, unterstreichen die bestehenden Probleme die Notwendigkeit, neue Wege zu gehen. Dazu werden aktuell verschiedene Ansätze diskutiert:
- Zulassungsbehörden sollten weniger verkürzte Entwicklungsprogramme akzeptieren, sondern zu robusten Evidenzanforderungen zurückkehren. Da die Entscheidungen über das Vorgehen von Zulassungsbehörden unmittelbare Auswirkungen auf Gesundheitssysteme haben, sollten alle Parteien, die Verantwortung für Gesundheitssysteme tragen, in strategische Entscheidungen im Zusammenhang mit der Zulassung eingebunden werden.
- Informationslücken zum Vergleich neuer Therapieoptionen mit Therapiestandards sollten durch die verpflichtende Durchführung aktiv kontrollierter Studien geschlossen werden. Die aktuelle Diskussion um eine europäische Regelung von HTA bietet eine Möglichkeit dazu.
- Erstattungs- und Preisbildungsentscheidungen sollten relevante Therapiefortschritte und eine sichere Entscheidungsgrundlage belohnen, nicht marginale Änderungen oder hoch unsichere Evidenz.
- Auf längere Sicht sollten die Verantwortlichen für die Gesundheitssysteme die Arzneimittelentwicklung aktiver gestalten. Dazu kann die Definition von relevanten Lücken in der Gesundheitsversorgung und die gezielte Förderung von Arzneimittelentwicklung in diesen Bereichen gehören. Entsprechende Ansätze existieren bereits für Antibiotika oder vernachlässigte Erkrankungen.
- Ein weiterer Vorschlag, die Effizienz, Qualität und Relevanz von Arzneimittelentwicklung zu verbessern, ist ein Open-Source-Model. Das Potenzial dieses Vorgehens wurde kürzlich von der EMA am Beispiel der Alzheimer-Erkrankung demonstriert und könnte anders als in diesem Beispiel über den Austausch von Informationen zwischen Zulassungsbehörden hinausgehen.
Fazit
Die aktuellen Ergebnisse der Arzneimittelentwicklung in Europa sind unzureichend. Anstrengungen auf nationaler und europäischer Ebene sollten Ziele definieren, die sich am Bedarf der Gesundheitssysteme orientieren, und auf dieser Basis Entwicklungs-, Zulassungs- und Bewertungsprozesse von Arzneimitteln neu gestalten.
Interessenkonflikte
Ein Interessenkonflikt wird von der Autorin verneint.
Literatur
- Wieseler B, McGauran N, Kaiser T: New drugs: where did we go wrong and what can we do better? BMJ 2019; 366: l4340.
vorab online
Dieser Artikel wurde am 15. Juli 2020 vorab online veröffentlicht.