Upadacitinib (Rinvoq®) ▼
Zugelassene Indikation und Wirkmechanismus
Upadacitinib (Rinvoq®) ist zur Behandlung der mittelschweren bis schweren aktiven rheumatoiden Arthritis (RA) bei Erwachsenen zugelassen, die auf ein oder mehrere krankheitsmodifizierende Antirheumatika (Disease-modifying antirheumatic drugs, DMARD) unzureichend angesprochen oder diese nicht vertragen haben. Das Arzneimittel kann als Monotherapie oder in Kombination mit Methotrexat (MTX) angewendet werden. DMARD werden in drei Untergruppen eingeteilt: konventionell synthetisch („conventional synthetic“, csDMARD) wie Hydroxychloroquin, Leflunomid, Methotrexat und Sulfasalazin; biologisch („biological“, bDMARD) wie z. B. Adalimumab, Certolizumab, Golimumab, Etanercept, Infliximab und Rituximab und auf spezifische Molekularstrukturen abzielend („targeted synthetic“, tsDMARD) wie Baricitinib und Tofacitinib.
Upadacitinib ist ein selektiver und reversibler Januskinasen(JAK)-Inhibitor (insbesondere von JAK1). Die JAK sind rezeptorassoziierte, intrazelluläre Enzyme, die an der Signalweiterleitung der Zytokine und Wachstumsfaktoren beteiligt sind, die viele zelluläre Prozesse wie u. a. Entzündungsreaktionen, Hämatopoese und Immunüberwachung steuern. Dadurch wirkt Upadacitinib immunsuppressiv und entzündungshemmend.
Markteinführung und frühe Nutzenbewertung
Rinvoq® (Upadacitinib) ist seit dem 1. Februar 2020 in dieser Indikation auf dem deutschen Markt.
Bewertung
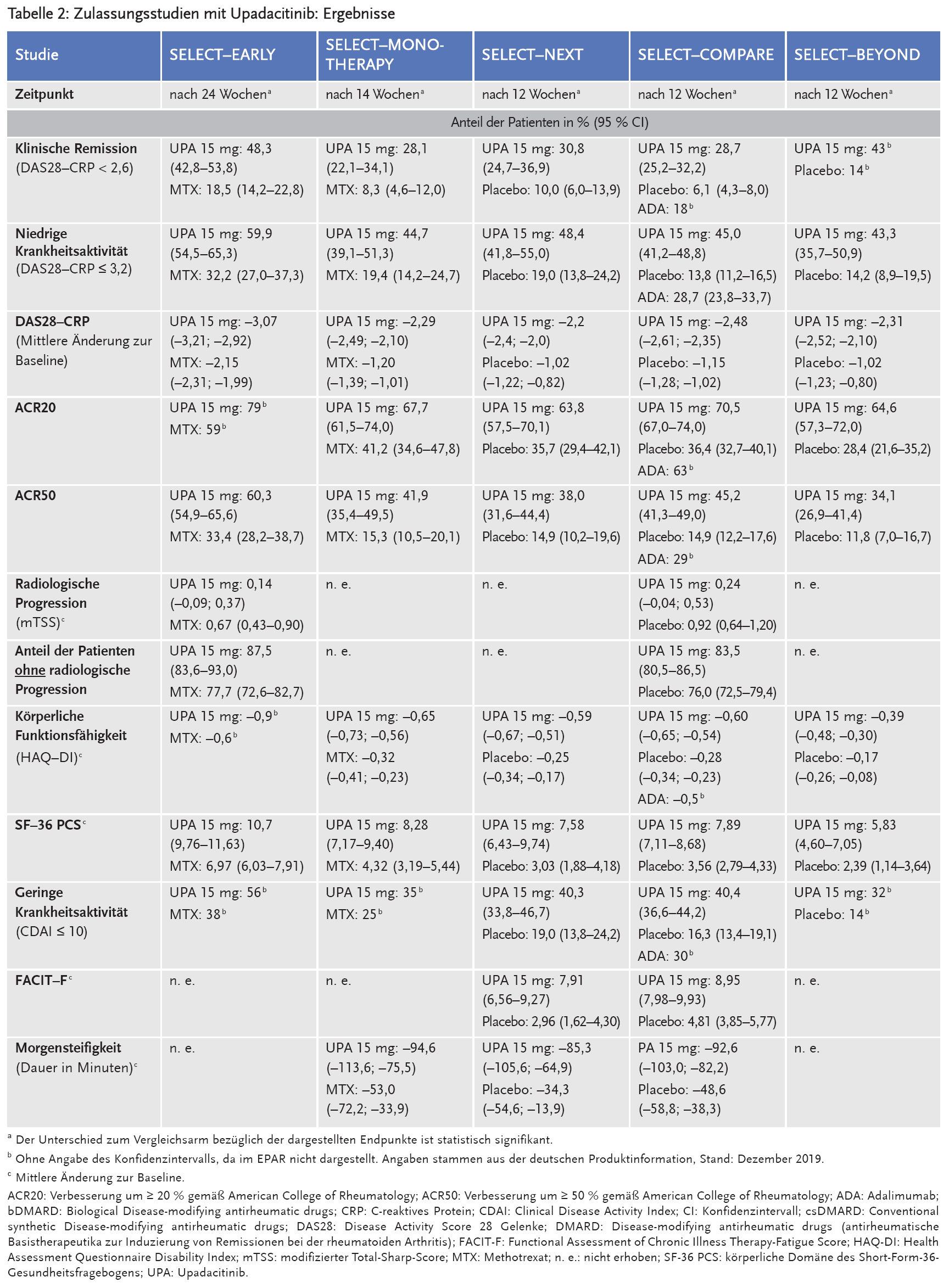
Unter der täglichen Gabe von Upadacitinib erreichen etwa 28–48 % der Patienten mit mittelschwerer bis schwerer aktiver rheumatoider Arthritis (RA) eine klinische Remission und etwa die Hälfte der Patienten eine niedrige Krankheitsaktivität. Auf eine First-line-Monotherapie mit Upadacitinib sprechen MTX-naive Patienten am besten an (48,3 % erreichen eine klinische Remission nach sechs Monaten), dafür wurde Upadacitinib allerdings nicht zugelassen. Patienten, die unzureichend auf die Behandlung mit csDMARD ansprechen bzw. unzureichendes Ansprechen oder Unverträglichkeit auf mindestens einem bDMARD zeigen, erreichen unter der Kombinationsbehandlung mit Upadacitinib und einem csDMARD zu 30,8 % eine klinische Remission nach zwölf Wochen. Dabei ist die Kombination Upadacitinib + csDMARD besser als Placebo unabhängig davon, welches csDMARD – Leflunomid, Chloroquin, Hydroxychloroquin oder Sulfasalazin – eingesetzt wurde. Es liegen allerdings keine Daten zu den Endpunkten in Abhängigkeit des eingesetzten csDMARD vor. Patienten mit unzureichendem Ansprechen auf MTX sprechen auf die Monotherapie im gleichen Ausmaß wie auf die Kombinationstherapie mit MTX und Upadacitinib an.
Upadacitinib bietet gegenüber den verfügbaren bDMARD den Vorteil der oralen Gabe, scheint aber genauso häufig schwere Infektionen als Nebenwirkung hervorzurufen. Zudem wurde eine Nichtunterlegenheit von Upadacitinib in Kombination mit MTX lediglich gegenüber Adalimumab bei Patienten mit unzureichendem Ansprechen auf MTX gezeigt.
Die Behandlung führt zu einem Anstieg der Lipidwerte (Gesamtcholesterin, LDL und HDL), die Auswirkung auf kardiovaskuläre Morbidität und Mortalität ist derzeit unklar, insbesondere weil Patienten mit RA ein erhöhtes Risiko für kardiovaskuläre Erkrankungen aufweisen. Bei Patienten, die mit Upadacitinib behandelt werden, sollten daher im Rahmen der Routinebehandlung Risikofaktoren (z. B. Hypertonie, Hyperlipidämie) bedacht werden.
Die Behandlung mit Upadacitinib kann zu einer Verschlimmerung einer bestehenden Infektion führen oder die Wahrscheinlichkeit für eine Infektion erhöhen. Die Langzeitsicherheit von Upadacitinib kann derzeit nicht abschließend bewertet werden.
Wirksamkeit in den Zulassungsstudien
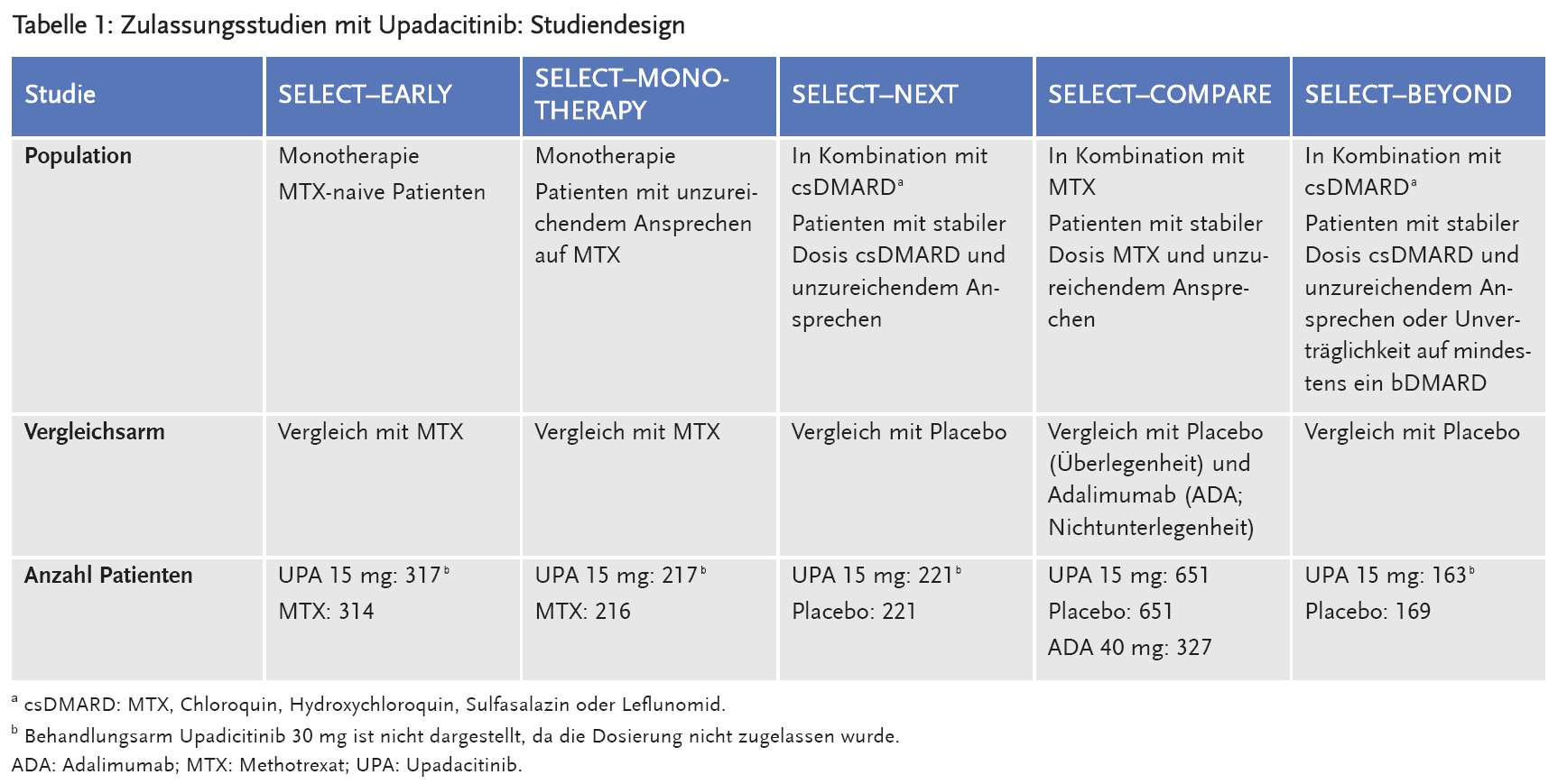
Für die Zulassung wurden fünf randomisierte, doppelblinde, multizentrische Phase-III-Studien bei Patienten mit mittelschwerer bis schwerer aktiver RA eingereicht. Eingeschlossen wurden Erwachsene, die mindestens sechs druckschmerzhafte und sechs geschwollene Gelenke aufwiesen. Zudem sollte der Nachweis einer systemischen Entzündung auf Basis der hsCRP-Erhöhung1 bzw. von Anti-CCP-Antikörpern vorliegen. Alle Studien bestehen aus einer Behandlungsphase von 48 Wochen und einer Langzeit-Fortsetzungsphase von vier bis fünf Jahren, in der die Patienten aus den Placebo-Armen bzw. aus den Vergleichsarmen mit unzureichendem Ansprechen auf Upadacitinib wechseln konnten. Die Patienten durften davor keinen JAK-Inhibitor (z. B. Tofacitinib, Baricitinib oder Filgotinib) bekommen haben. In den Studien wurde Upadacitinib in der Dosierung 15 mg oder 30 mg einmal täglich untersucht. Zugelassen wurde allerdings nur die Dosierung von 15 mg einmal täglich, weil sich unter der 30-mg-Dosierung nur marginale Zusatzeffekte bezüglich der Wirksamkeit zeigten. Das Studiendesign ist in Tabelle 1 dargestellt.
Primärer Endpunkt der Studien war entweder die klinische Remission, definiert als DAS28–CRP < 2,6 (SELECT–EARLY und SELECT–COMPARE), oder eine niedrige Krankheitsaktivität, definiert als DAS28–CRP ≤ 3,2 (SELECT–NEXT, SELECT–MONOTHERAPY und SELECT–BEYOND). Als sekundäre Endpunkte wurden u. a. die Verbesserung um ≥ 20 % (oder ≥ 50 %) gemäß American College of Rheumatology (ACR20, ACR50), die radiologische Progression anhand des modifizierten Total-Sharp-Scores (mTSS) und geringe Krankheitsaktivität anhand des Clinical Disease Activity Index (CDAI). Patient-Reported Outcomes (PRO) wurden erhoben anhand des Health Assessment Questionnaire Disability Index (HAQ-DI)2 zur Messung von körperlicher Funktion und Behinderung, der Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F)3 zur Beurteilung von Müdigkeit sowie der körperlichen Domäne des Short-Form-36-Gesundheitsfragebogens (SF-36 PCS)4 als Maß für die Lebensqualität. Die Ergebnisse der Zulassungsstudien sind für die Dosis 15 mg Upadacitinib täglich in Tabelle 2 dargestellt. Die Ergebnisse des Behandlungsarms 30 mg Upadicitinib 30 mg täglich sind nicht dargestellt, da die Dosierung nicht zugelassen wurde.
Einer aktuellen Bayes-Netzwerk-Metaanalyse zur Kombination direkter und indirekter Evidenz aus randomisierten kontrollierten Studien bei Patienten mit aktiver RA zufolge wurde die höchste ACR20-Ansprechrate mit der Kombination Upadacitinib 15 mg + MTX erreicht. Dieser folgten Upadacitinib 30 mg + MTX, Adalimumab 40 mg + MTX, Upadacitinib 30 mg und Upadacitinib 15 mg als Monotherapie sowie MTX allein. Die Sicherheit wurde anhand der Anzahl schwerer Nebenwirkungen evaluiert und unterschied sich nicht signifikant zwischen den sechs Interventionen. Die Autoren schlussfolgern daher, dass Upadacitinib 15 mg (bzw. 30 mg) einmal täglich in Kombination mit MTX (einmal wöchentlich) die wirksamste Intervention bei aktiver RA ist, die kein erhöhtes Risiko für schwere Nebenwirkungen mit sich bringt5.
Ausgewählte Nebenwirkungen
Sehr häufig treten Infektionen der oberen Atemwege auf, häufig Neutropenie, Hypercholesterinämie, Husten, Übelkeit, Fieber, Anstieg der Lebertransaminasen. Anstieg der Kreatinkinase und Gewichtserhöhung. Gelegentliche Nebenwirkungen sind Pneumonien, Herpes zoster- und Herpes simplex-Infektionen sowie orale Candidosen und Hypertriglyzeridämie.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Die empfohlene Dosis von Upadacitinib beträgt 15 mg einmal täglich oral mit oder unabhängig von einer Mahlzeit. Die mittlere terminale Eliminationshalbwertszeit von Upadacitinib beträgt 9 bis 14 Stunden.
- Die Behandlung sollte bei Patienten mit einer absoluten Lymphozytenzahl < 500 Zellen/mm3, einer absoluten Neutrophilenzahl < 1000 Zellen/mm3 oder einem Hämoglobinspiegel < 8 g/dl nicht begonnen werden bzw. unterbrochen werden, bis sich die Abweichungen normalisiert haben.
- Wenn bei einem Patienten eine schwere Infektion auftritt, sollte die Behandlung unterbrochen werden, bis die Infektion unter Kontrolle ist.
- Für Patienten ab 75 Jahren sowie für Patienten mit schwerer Niereninsuffizienz (eGFR: 15–29 ml/min/1,73 m2) liegen nur begrenzte Erfahrungen vor.
- Eine Dosisanpassung ist weder bei Patienten mit leichter oder mittelschwerer Niereninsuffizienz noch bei Patienten mit leichter (Child-Pugh A) oder mittelschwerer (Child-Pugh B) Leberinsuffizienz erforderlich. Upadacitinib darf bei Patienten mit schwerer Leberinsuffizienz (Child-Pugh C) nicht angewendet werden.
- Upadacitinib sollte nur nach Nutzen-Risiko-Abwägung angewendet werden bei Patienten mit chronischen oder wiederkehrenden Infektionen; Exposition gegenüber Tuberkulose (z. B. Leben oder Reisen in Gebieten mit endemischer Tuberkulose oder endemischen Mykosen); schweren oder opportunistischen Infektion in der Anamnese oder Grunderkrankungen, aufgrund derer sie anfällig für Infektionen sind.
- Upadacitinib wird hauptsächlich durch CYP3A4 metabolisiert, daher kann seine Plasmaexposition durch Arzneimittel beeinflusst werden, die CYP3A4 stark hemmen (wie z. B. Ketoconazol, Itraconazol, Posaconazol, Voriconazol und Clarithromycin) oder induzieren (wie z. B. Rifampicin und Phenytoin).
- Upadacitinib sollte bei Patienten mit hohem Risiko für tiefe Beinvenenthrombosen (TVT) oder Lungenembolien (LE) (z. B. höheres Alter, Adipositas, TVT/LE in der Anamnese, größere Operationen oder längere Immobilisierung) mit Vorsicht angewendet werden.
- Die Anwendung von attenuierten Lebendimpfstoffen während oder unmittelbar vor einer Behandlung mit Upadacitinib wird nicht empfohlen.
- Die Anwendung in der Schwangerschaft ist kontraindiziert, bei Frauen in gebärfähigem Alter ist eine wirksame Empfängnisverhütung unabdingbar.
Schulungsmaterial
Für einzelne Arzneimittel wird bereits bei der Zulassung angeordnet, dass das Arzneimittel nur unter Verwendung von Schulungsmaterialien in Verkehr gebracht werden darf. Das Schulungsmaterial dient dazu, die Wissensvermittlung zu optimieren und Hilfe bei der sicheren Anwendung des Arzneimittels zu geben, gegebenenfalls unter Einbeziehung einer patientenbezogenen Ansprache. Das behördlich beauflagte und genehmigte Schulungsmaterial zu Upadacitinib beinhaltet eine Informationsbroschüre für den Arzt sowie einen Patientenpass.

Weiterführende Informationen
Das IQWiG wurde am 01.02.2020 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Rinvoq®, erschienen am 18. Dezember 2019. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Fußnoten
1Hochsensitives (High-sensitivity) C-reaktives Protein als Parameter zur Abschätzung der Krankheitsaktivität bei RA.
2HAQ-DI: 0 = bester Wert; 3 = schlechtester Wert; 20 Fragen; 8 Kategorien: Ankleiden und Körperpflege, Aufstehen, Essen, Gehen, Hygiene, Greifen, Festhalten und andere Tätigkeiten.
3FACIT-F: 27 Items zur Bewertung von Lebensqualität chronisch Erkrankter in den vier Bereichen körperliches, emotionales, funktionelles und soziales Wohlbefinden. Höhere Werte indizieren bessere Lebensqualität.
4SF-36 PCS: acht Domänen – Vitalität, körperliche Funktionsfähigkeit, körperliche Schmerzen, allgemeine Gesundheitswahrnehmung, körperliche Rollenfunktion, emotionale Rollenfunktion, soziale Funktionsfähigkeit, psychisches Wohlbefinden; je höher die Punktzahl, desto geringer die Behinderung; 0 = maximale Behinderung, 100 = keine Behinderung.
5Song GG, Lee YH: Comparative efficacy and safety of 15 and 30 mg upadacitinib administered to patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Z Rheumatol 2020; 79: 103-111.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 25. März 2020 vorab online veröffentlicht.