Monoklonale Antikörper zur Prophylaxe von Migräne: Wirksamkeit und Stellenwert
Zusammenfassung
Migräne ist eine häufige Ursache für vorübergehende körperliche Einschränkungen und zudem mit anderen Erkrankungen assoziiert. Seit 2018 wurden drei monoklonale Antikörper zur Prophylaxe von Migräne bei Erwachsenen mit mindestens vier Migränetagen pro Monat zugelassen. Der Artikel gibt eine Übersicht über ihre Wirksamkeit und Sicherheit sowie über die frühe Nutzenbewertung und die Jahrestherapiekosten, um Ärzten eine Hilfestellung zur evidenzbasierten und rationalen Verordnung von Arzneimitteln zur Migräneprophylaxe zu geben.
Migräne
Für Kopfschmerzen besteht eine Lebenszeitprävalenz von etwa 66 %; 14,4 % davon entfallen auf Migräne. Die Prävalenz ist bei Frauen (18,9 %) zweimal höher als bei Männern (9,8 %) (1). Obwohl nur 6 % der Männer betroffen sind, ist Migräne eine der häufigsten neurologischen Erkrankungen bei Männern. Migräne ist einerseits eine häufige Ursache für vorübergehende körperliche Einschränkungen und andererseits aber auch mit anderen Erkrankungen wie beispielsweise Depression, Epilepsie und vaskulären Erkrankungen assoziiert (2).
Die deutsche S1-Leitlinie „Therapie der Migräneattacke und Prophylaxe der Migräne“ leitet die Indikation zu einer medikamentösen Prophylaxe der Migräne aus dem besonderen Leidensdruck, der Einschränkung der Lebensqualität und dem Risiko eines Medikamentenübergebrauchs ab. Ziel der medikamentösen Prophylaxe ist die Reduktion von Häufigkeit, Schwere und Dauer der Migräneattacken und die Prophylaxe des Kopfschmerzes bei Übergebrauch von Schmerz- und Migränemitteln. Eine Wirksamkeit einer Migräneprophylaxe wird bei einer Reduktion der Anfallshäufigkeit von 50 % oder mehr angenommen (3).
Zur Migräneprophylaxe werden in Deutschland eingesetzt: Betablocker (Metoprolol, Propranolol), Amitriptylin, Flunarizin, Topiramat, Valproinsäure und Clostridium botulinum Toxin Typ A (Botulinumtoxin Typ A). Aufgrund der jeweiligen Zulassung der Arzneimittel kommen für nicht vorbehandelte Patienten die Wirkstoffe Metoprolol, Propranolol, Flunarizin, Topiramat oder Amitriptylin als Mittel der ersten Wahl infrage. Zwischen diesen Wirkstoffen besteht keine klare, evidenzbasierte Präferenz. Gleichwohl ist z. B. Flunarizin nur zugelassen, wenn die Behandlung mit Betablockern kontraindiziert ist oder keine ausreichende Wirkung gezeigt hat (4), und Topiramat nur nach sorgfältiger Abwägung möglicher alternativer Behandlungsmethoden (5). Wenn diese First-line-Wirkstoffe ohne Erfolg bleiben oder nicht geeignet sind, z. B. aufgrund von Kontraindikationen, können Valproinsäure oder Botulinumtoxin Typ A eingesetzt werden. Botulinumtoxin Typ A ist bei chronischer Migräne bei Patienten zugelassen, die auf prophylaktische Migränebehandlung nur unzureichend angesprochen oder diese nicht vertragen haben (6). Valproinsäure darf zur Migräneprophylaxe bei Erwachsenen ab 18 Jahren im Rahmen des Off-Label-Use nur eingesetzt werden, wenn eine Behandlung mit anderen dafür zugelassenen Arzneimitteln nicht erfolgreich war oder kontraindiziert ist (7).
Seit Mitte 2018 wurden in der EU bereits drei monoklonale Antikörper (monoclonal antibodies, mAbs) zur Prophylaxe von Migräne bei Erwachsenen mit mindestens vier Migränetagen pro Monat zugelassen: Erenumab (Aimovig®) wurde im Juli 2018 zugelassen, im November 2018 folgte die Zulassung von Galcanezumab (Emgality®) und im März 2019 von Fremanezumab (Ajovy®) (8).
Drei monoklonale Antikörper zur Migräneprophylaxe
Die drei monoklonalen Antikörper werden gentechnisch in Ovarialzellen chinesischer Hamster (Chinese hamster ovary, CHO) hergestellt. Sie richten sich spezifisch gegen das migräneauslösende Neuropeptid Calcitonin-Gene-Related-Peptide (CGRP) und haben damit ein neues Wirkprinzip, das an einem für die Pathophysiologie der Migräne zentralen Mechanismus angreift. CGRP reguliert die nozizeptive Signalübertragung und wirkt als Vasodilatator. Der CGRP-Spiegel steigt – im Gegensatz zu anderen Neuropeptiden – während eines Migräneanfalls an und normalisiert sich beim Abklingen der Kopfschmerzen. Die intravenöse Infusion von CGRP löst bei Patienten migräneähnliche Kopfschmerzen aus. Der CGRP-Rezeptor findet sich gehäuft in Regionen, die für die Pathophysiologie der Migräne relevant sind, wie etwa dem Ganglion trigeminale (9).
Erenumab bindet mit hoher Affinität und Spezifität an den CGRP-Rezeptor und hemmt damit den CGRP-Signalweg. Galcanezumab und Fremanezumab binden an CGRP und hindern dieses an der Bindung und somit an der Aktivierung seines Rezeptors.
Die Arzneimittel sind als Fertigpen verfügbar und werden subkutan am Abdomen, am Oberschenkel, an der Außenseite des Oberarms oder in den Gesäßbereich appliziert. Eine intravenöse oder intramuskuläre Applikation darf nicht erfolgen. Werden wiederholte Injektionen verabreicht, sollte die Injektionsstelle gewechselt werden. Patienten sollen nach angemessener Schulung die Arzneimittel selbst verabreichen können. Die Arzneimittel müssen im Kühlschrank bei 2–8°C im Umkarton gelagert werden, um den Inhalt vor Licht zu schützen. Zur Vermeidung von Beschwerden an der Injektionsstelle sollte das Medikament vor der Injektion mindestens 30 Minuten bei Raumtemperatur (bis zu 25 °C) belassen werden. Wird eine Injektion am geplanten Termin versäumt, sollte die Verabreichung so bald wie möglich mit der angezeigten Dosis gemäß Dosierungsplan wieder aufgenommen werden.
Bewertung
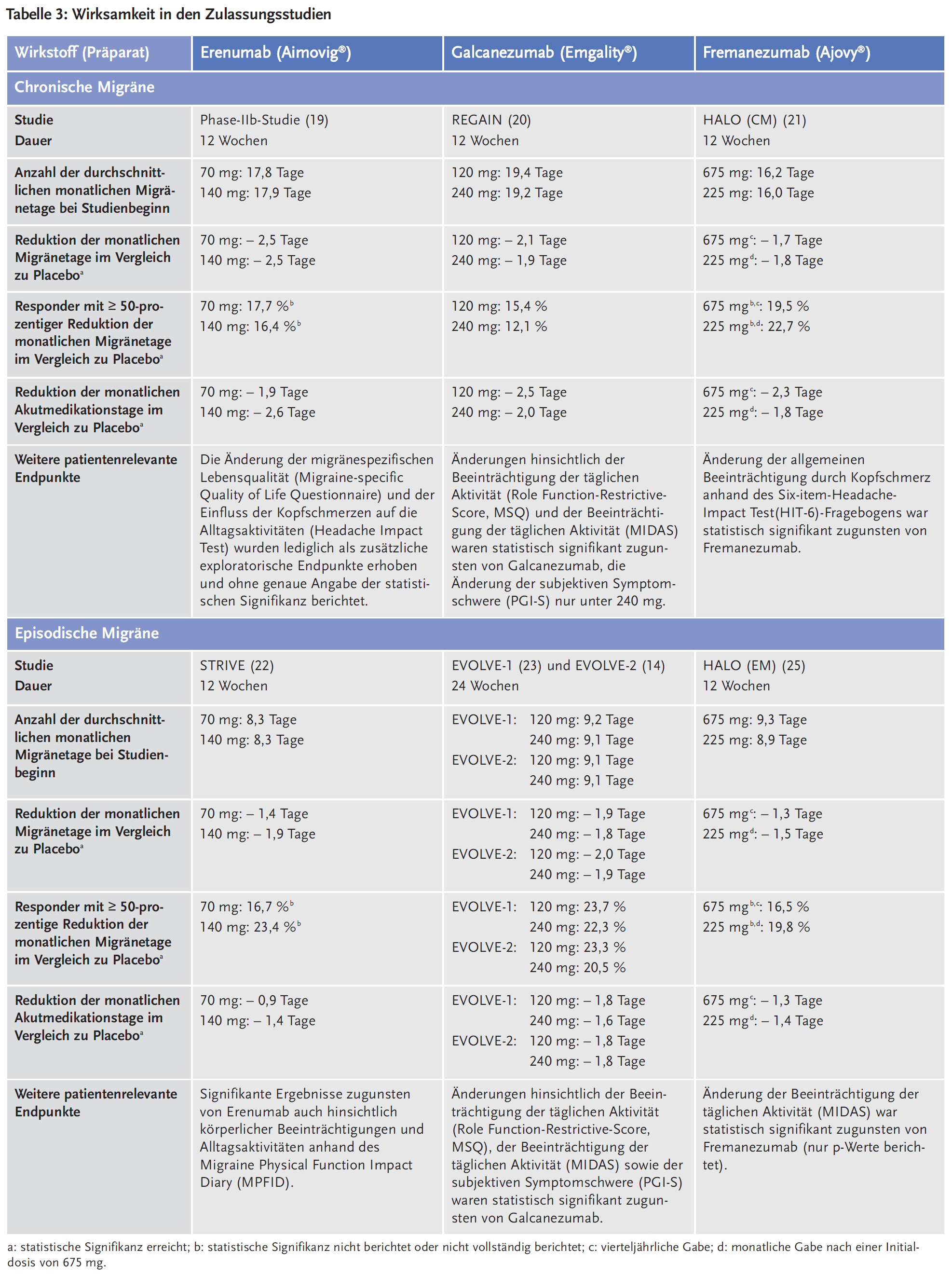
Erenumab, Galcanezumab und Fremanezumab haben ein neues Wirkprinzip, welches an einem für die Pathophysiologie der Migräne zentralen Mechanismus angreift. Allerdings ist ihre prophylaktische Wirksamkeit lediglich als moderat einzustufen, ähnlich den bisher verfügbaren Arzneimitteln zur Migräneprophylaxe. Auch wenn Responderanalysen zum Anteil der Patienten mit einer mindestens 50-prozentigen Reduktion der Migränetage pro Monat, die Wirksamkeit der Antikörper relativ hoch erscheinen lassen, betrug in den Zulassungsstudien die absolute Reduktion der monatlichen Migränetage gegenüber Placebo zwischen 1,2 und 2,3 Tage bei EM und zwischen 1,7 und 2,5 Tage bei CM (vgl. Tabelle 3). Damit ist der „Gewinn“ für die Patienten überschaubar, wenn man den Placeboeffekt berücksichtigt: Nur jeder fünfte bis sechste Patient hat durch einen der mAbs mehr als 50 % weniger Migränetage pro Monat. Dafür beträgt die Number needed to treat (NNT) im Vergleich zu Placebo 4–8 bei CM und 4–6 bei EM. Aus klinischer Sicht entspricht dies einem moderaten therapeutischen Nutzen.
Ein Vorteil gegenüber bisher verfügbaren Wirkstoffen scheint die bessere Verträglichkeit zu sein. Ein weiterer Vorteil könnte die vierwöchentliche bzw. die vierteljährliche Applikation (bei Fremanezumab) sein, die allerdings subkutan erfolgen muss. Nach derzeitiger Datenlage scheint die gute Verträglichkeit, speziell das Fehlen sedierender, metabolischer, kognitiver und depressionsauslösender Eigenschaften einen Vorteil gegenüber anderen prophylaktischen Maßnahmen (Topiramat, Betablocker, Amitriptylin) darzustellen.
Es liegen keine direkten Vergleiche der drei verfügbaren Antikörper Erenumab, Galcanezumab und Fremanezumab vor. CGRP hat eine ausgeprägte vasodilatatorische Wirkung. Die Hemmung seines Rezeptors birgt daher theoretisch das Risiko für kardiovaskuläre Ereignisse, das bei Migräne ohnehin gering erhöht ist. Die verfügbaren Studien ergaben keine eindeutigen Hinweise auf ein erhöhtes kardiovaskuläres Risiko, allerdings wurden vorbelastete Patienten ausgeschlossen. Die Risiken einer langfristigen Blockade von CGRP können zum jetzigen Zeitpunkt nicht abschließend beurteilt werden, da Langzeitdaten fehlen.
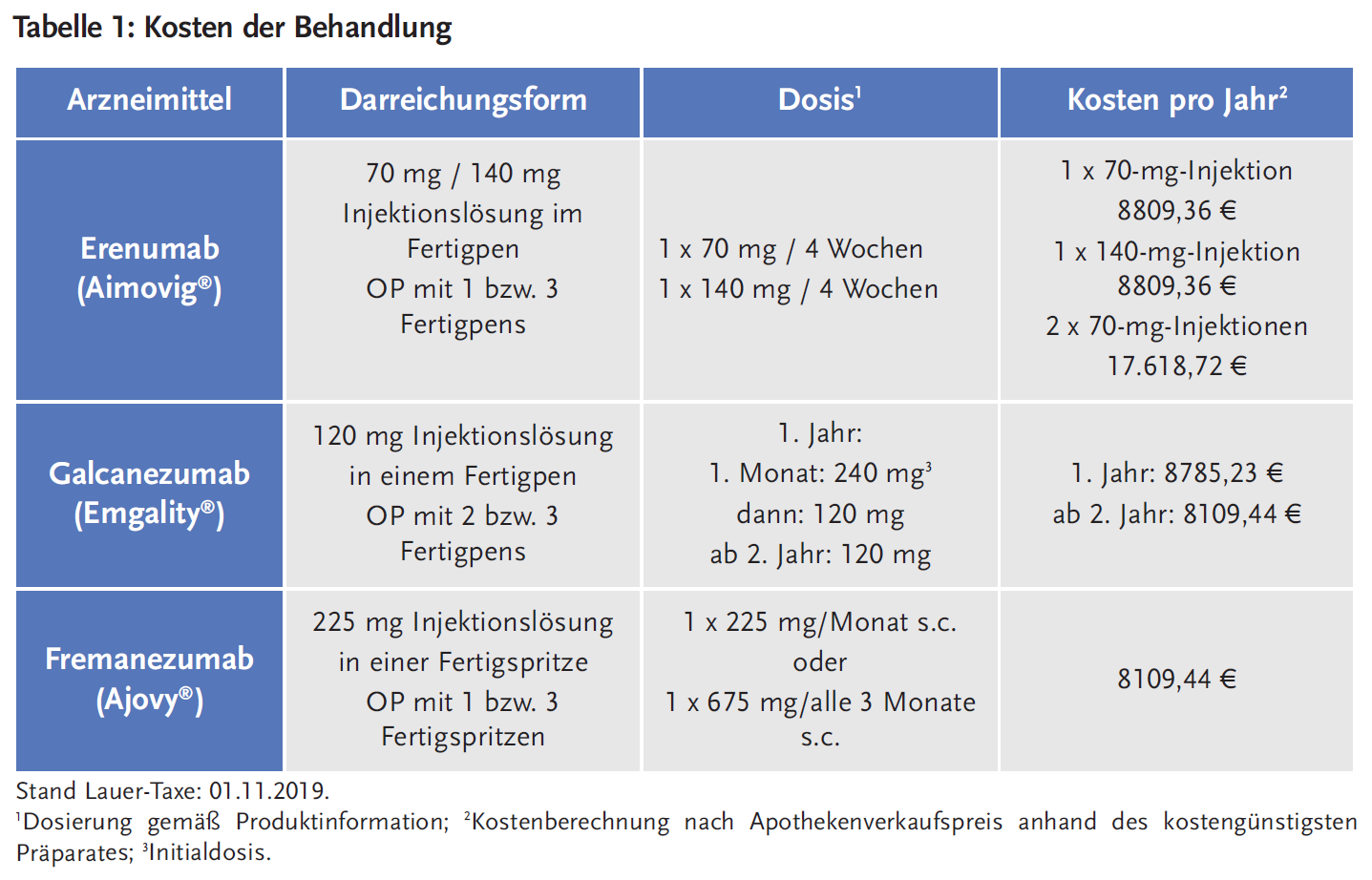
Der Einsatz von Erenumab, Galcanezumab und Fremanezumab sollte daher vorerst nur nach Versagen anderer Arzneimittel zur Migräneprophylaxe oder bei deren Unverträglichkeit erfolgen. Zudem sind bei der Verordnung dieser Arzneimittel die Kosten der Behandlung berücksichtigt werden, die in Tabelle 1 dargestellt sind. Derzeit sind die Jahrestherapiekosten am niedrigsten bei Fremanezumab, wobei zu berücksichtigen ist, dass die Erstattungsbeträge nach § 130b SGB V für die drei Arzneimittel derzeit noch verhandelt werden (10).
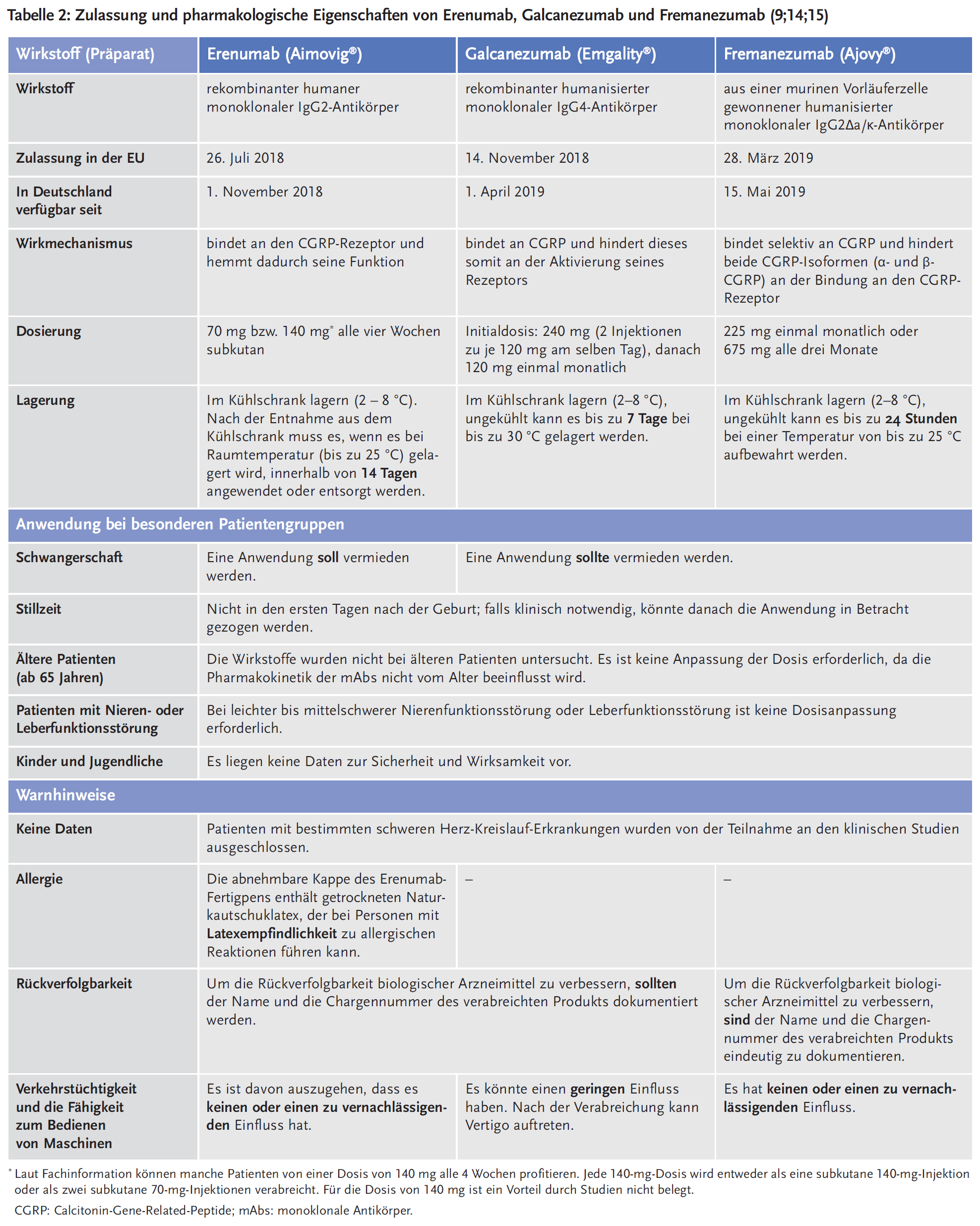
Pharmakologische Eigenschaften und Anwendung bei besonderen Patientengruppen
Für die Zulassung der Antikörper wurden keine Wechselwirkungsstudien durchgeführt. Basierend auf den Eigenschaften der Wirkstoffe sind keine pharmakokinetischen Wechselwirkungen zu erwarten. Aufgrund der Metabolisierungswege monoklonaler Antikörper wird keine Wirkung auf gleichzeitig verabreichte andere Arzneimitteln erwartet. Darüber hinaus ergab sich während der klinischen Zulassungsstudien bei der gleichzeitigen Anwendung von Arzneimitteln zur Akutbehandlung der Migräne (insbesondere Analgetika, Ergotaminderivate und Triptane) oder zur Vorbeugung der Migräne keine Beeinträchtigung der Pharmakokinetik von Erenumab, Galcanezumab und Fremanezumab.
Es wurden keine spezifischen pharmakologischen Studien zur Beurteilung der Auswirkungen von Nierenfunktionsstörungen und Leberfunktionsstörungen auf die Pharmakokinetik der neuen Wirkstoffe durchgeführt. Die renale Elimination monoklonaler IgG-Antikörper ist gering; zudem werden sie hauptsächlich über einen intrazellulären Katabolismus eliminiert. Daher ist auch nicht zu erwarten, dass eine Leberinsuffizienz die Clearance der monoklonalen Antikörper beeinflusst. Bei Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung oder Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Patienten mit schwerer Leberfunktionsstörung oder Nierenfunktionsstörung (eGFR < 30 ml/min/1,73 m2) wurden in den Studien nicht untersucht.
Es liegen nur begrenzte Erfahrungen mit der Anwendung von Erenumab, Galcanezumab und Fremanezumab bei Schwangeren vor. Zwar haben tierexperimentelle Studien keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität ergeben, eine Anwendung während der Schwangerschaft soll allerdings laut Fachinformation aus Vorsichtsgründen vermieden werden.
Es ist nicht bekannt, ob Erenumab, Galcanezumab oder Fremanezumab in die menschliche Muttermilch übergehen. Humane IgG gehen in den ersten Tagen nach der Geburt in die Muttermilch über, ihre Konzentration sinkt danach auf niedrige Werte ab. Ein Risiko für das gestillte Kind während dieser Zeitspanne kann nicht ausgeschlossen werden. Falls es klinisch notwendig ist, könnte danach die Anwendung der Antikörper während der Stillzeit in Betracht gezogen werden.
Patienten mit schweren kardiovaskulären Erkrankungen waren von einer Teilnahme an den klinischen Studien ausgeschlossen: u. a. Patienten mit vorbestehendem Myokardinfarkt, Schlaganfall, transitorischen ischämischen Attacken, instabiler Angina pectoris, koronarer arterieller Bypass-Operation oder anderen durchgeführten Revaskularisierungsverfahren innerhalb der letzten zwölf Monaten vor dem Screening sowie Patienten mit schlecht kontrolliertem Bluthochdruck oder BMI > 40 kg/m2. Auch Patienten mit Opioid-Übergebrauch oder mit Arzneimittelübergebrauch wurden nicht eingeschlossen. Für diese Patientengruppe liegen keine Sicherheitsdaten zur Anwendung von Erenumab, Galcanezumab und Fremanezumab vor (11-13).
Zulassungsstudien
Alle drei Antikörper wurden jeweils in zwei internationalen, multizentrischen, doppelblinden, randomisierten, placebokontrollierten Phase-III-Studien an erwachsenen Patienten mit episodischer (EM) und chronischer Migräne (CM) untersucht. Bei Galcanezumab wurde zudem eine weitere Zulassungsstudie bei EM durchgeführt, um ein zweites Dosierungsschema zu evaluieren. Die Studiendauer betrug 12 Wochen bzw. 24 Wochen. Die rekrutierten Patienten wiesen eine mindestens zwölfmonatige Vorgeschichte von Migräne (mit und ohne Aura) auf, gemäß den Diagnosekriterien der internationalen Kopfschmerzklassifikation (International Classification of Headache Disorders, ICHD-III). Eine Beschreibung der Studien findet sich unter: Erenumab; Galcanezumab und Fremanezumab. Die Ergebnisse zu relevanten Endpunkten sind in Tabelle 3 zusammengestellt (11-13).
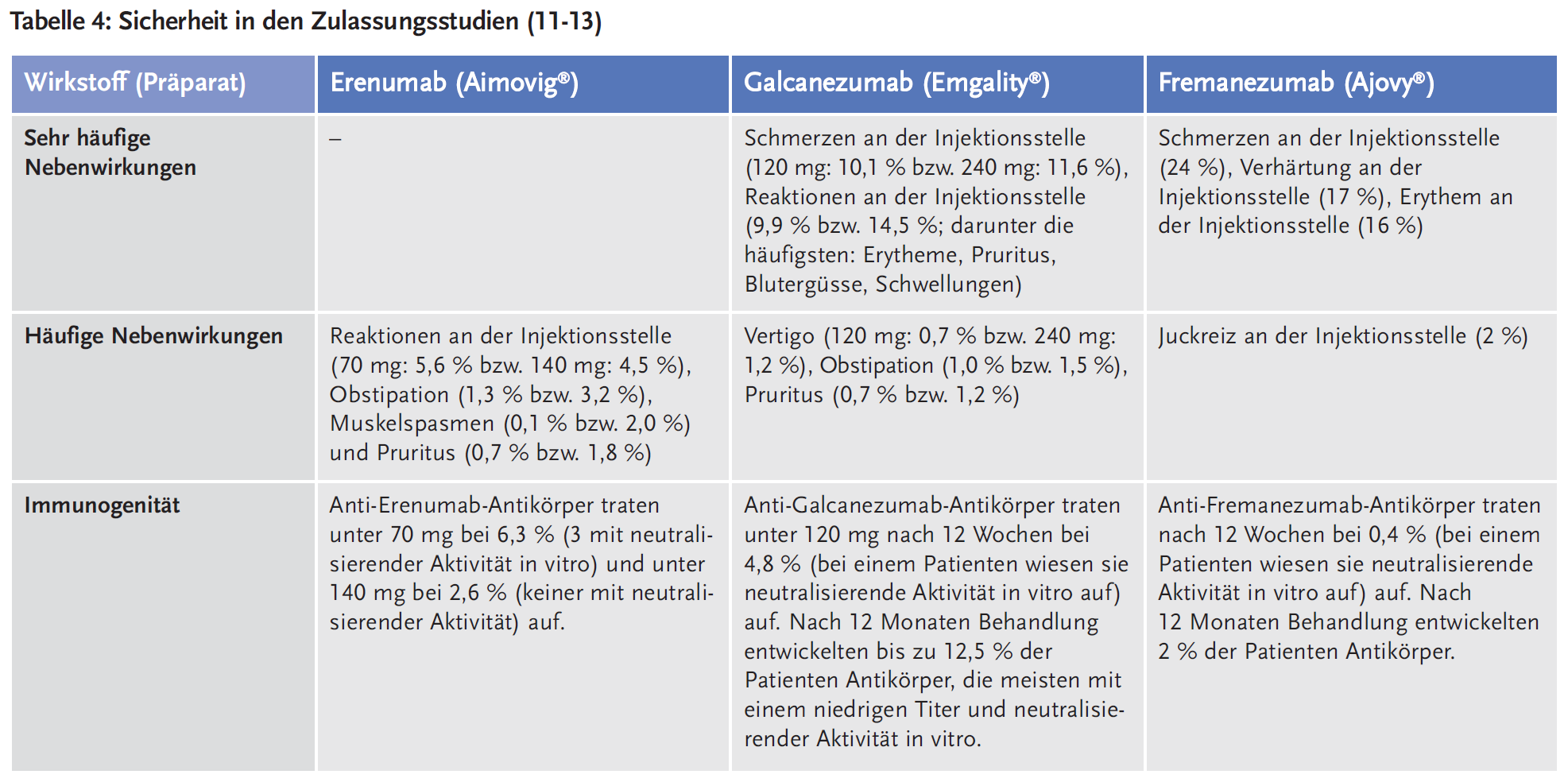
Die Nebenwirkungen, die während der doppelblinden Behandlungsphase der Zulassungsstudien auftraten und Eingang in die Fachinformationen fanden, sind in Tabelle 4 dargestellt. Zur Beurteilung der langfristigen Sicherheit der Antikörper sind die Daten aus den Zulassungsstudien allerdings nicht ausreichend, insbesondere weil ein theoretisches Risiko speziell für Patienten mit einem kardiovaskulären Risiko besteht, da für CGRP eine vasodilatatorische bzw. kontraktionshemmende Wirkung beschrieben wurde. Erenumab, Galcanezumab und Fremanezumab wirken als CGRP-Antagonisten potenziell vasokonstriktorisch. Damit verbundene mögliche Gefahren potenzieller kardiovaskulärer Risiken können naturgemäß erst nach längerer und breiterer Anwendung identifiziert werden, zumal in allen bisherigen Studien ältere und kardial vorbelastete Patienten weitgehend ausgeschlossen wurden. Da CGRP nicht nur ein Vasodilatator ist, sondern auch die Neovaskularisation und die Lymphangiogenese fördert, kann die langfristige CGRP-Blockierung möglicherweise auch die Erholung von Ischämien sowie die Heilung von Wunden und Ulzera behindern (16).
Migräne betrifft häufig Frauen im gebärfähigen Alter, daher verdienen die teratogenen Risiken besondere Beachtung. CGRP scheint in der Schwangerschaft für die Entwicklung und Anpassung der Gefäßsysteme im Uterus, in der Plazenta und im Fötus eine besondere Rolle zu spielen (17). Im Rattenversuch führte die Gabe eines CGRP-Antagonisten in der Schwangerschaft dosisabhängig zu einem erhöhten Blutdruck, zu geringerem Plazenta- und Geburtsgewicht und zu einer erhöhten fetalen Mortalität (18). Daher kann auch der mögliche Schaden durch Erenumab, Galcanezumab und Fremanezumab nicht abschließend beurteilt werden, solange keine umfangreichen Registerdaten über Schwangerschaftsverläufe unter der Therapie mit diesen Wirkstoffen vorliegen.
Meldung von Nebenwirkungen
Spontanmeldungen von Nebenwirkungen sind eines der wichtigsten Instrumente, um Risiken von Arzneimitteln frühzeitig zu erfassen. Insbesondere bei Arzneimitteln mit neuartigem Wirkmechanismus sind solche Meldungen essenziell. Sie tragen dazu bei, unbekannte Nebenwirkungen und wichtige Sicherheitssignale zu identifizieren, um das Sicherheitsprofil des Arzneimittels zu evaluieren und sein Risiko-Nutzen-Verhältnis zu bewerten. Nebenwirkungen unter der Behandlung mit Erenumab, Galcanezumab und Fremanezumab sollten gemeldet werden, um die Arzneimittelsicherheit dieser Wirkstoffe zu überwachen (https://www.akdae.de/Arzneimittelsicherheit/UAW-Meldung/).
Frühe Nutzenbewertung nach § 35a SGB V
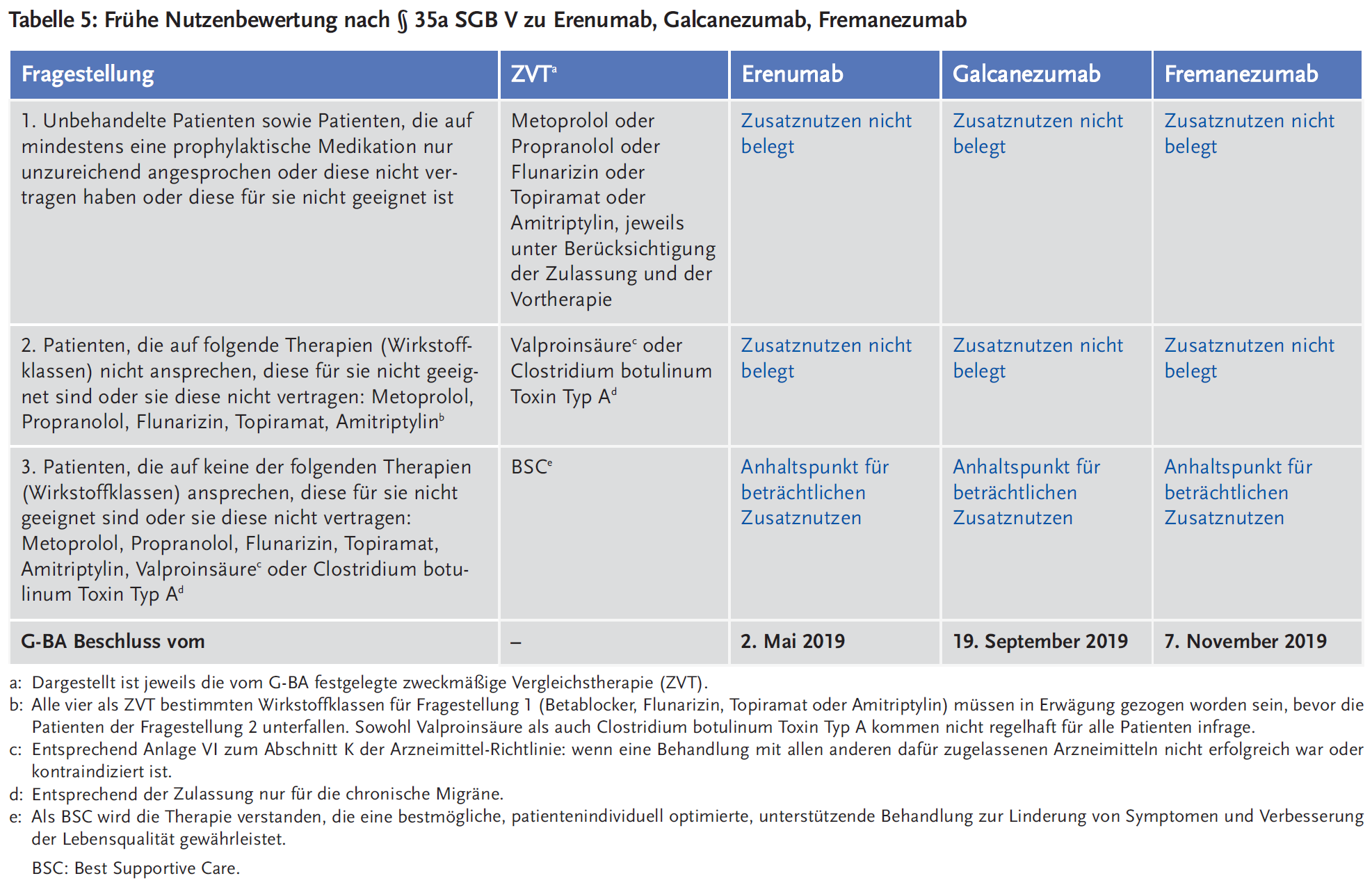
Die drei Antikörper – Erenumab, Galcanezumab und Fremanezumab – wurden als Arzneimittel mit neuen Wirkstoffen nach § 35a SGB V der frühen Nutzenbewertung in Deutschland unterzogen. Aufgrund der Zulassung der verfügbaren Therapieoptionen, die als zweckmäßige Vergleichstherapie in Frage kommen, bildete der Gemeinsame Bundesausschuss (G-BA) drei Patientenunterpopulationen, in denen der Zusatznutzen der Antikörper zu bewerten ist. Diese sind in Tabelle 5 dargestellt.
Zusammenfassend erkennt der G-BA einen Zusatznutzen der CGRP-Antikörper nur für hochgradig therapieresistente Migränepatienten. Ob Erenumab, Galcanezumab und Fremanezumab auch für bislang unbehandelte oder mit wenigen Substanzen vorbehandelte Patienten einen Zusatznutzen bringen, muss noch in kontrollierten Studien geprüft werden.
Erenumab
Zu Erenumab legte der pharmazeutische Unternehmer Daten nur zu Fragestellung 3 vor, also zu den hochgradig therapierefraktären Patienten. Die Bewertung des Zusatznutzens erfolgte auf Basis der randomisierten, doppelblinden Phase-III-Studie LIBERTY zum Vergleich von 140 mg Erenumab + Best Supportive Care (BSC) mit Placebo + BSC über einen Zeitraum von 12 Wochen bei erwachsenen Patienten mit EM (26). Die LIBERTY-Studie schloss 246 Patienten ein, die durchschnittlich 4 bis 14 Migränetage pro Monat (im Mittel 9,1 Migränetage pro Monat) innerhalb der letzten 3 Monate und einem Therapieversagen auf 2 bis 4 vorangegangene medikamentöse Migräneprophylaxen aufwiesen. Der primäre Endpunkt der Studie war der Anteil der Patienten mit einer Reduktion der Migränetage pro Monat um ≥ 50 % zu Woche 12.
Für die Nutzenbewertung wurde nur ein Teil der Patienten berücksichtigt, der aufgrund der Anzahl der Vortherapien die Charakteristika der Patientenpopulation für Fragestellung 3 erfüllte (n = 193 Patienten). Dabei zeigte sich für den Endpunkt Reduktion der Migränetage pro Monat um ≥ 50 % ein statistisch signifikanter, beträchtlicher Vorteil für Erenumab: 30 % versus 14 % (RR 2,25 [95 % Konfidenzintervall 1,25–4,03]; p = 0,005). Des Weiteren bestanden statistisch signifikante, klinisch relevante Effekte bezüglich der Aktivitätsbeeinträchtigung und der gesundheitsbezogenen Lebensqualität zugunsten von Erenumab, während sich bezüglich der Nebenwirkungen keine Unterschiede ergaben. Der G-BA stufte diese Effekte als bisher nicht erreichte deutliche Verbesserung des therapierelevanten Nutzens ein und damit den Zusatznutzen in dieser Subgruppe als beträchtlich. Da aber Unklarheiten bezüglich der Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext bestanden sowie Unsicherheit, weil in der Studie keine klare Abgrenzung zwischen EM und CM erfolgte, wurde die Aussagesicherheit der Daten als Anhaltspunkt bewertet (27).
Galcanezumab
Auch zu Galcanezumab legte der pharmazeutische Unternehmer Daten nur zu hochgradig therapierefraktären Patienten vor. Die Bewertung des Zusatznutzens erfolgte auf Basis der Zulassungsstudien EVOLVE-1 und EVOLVE-2 (bei EM) und REGAIN (bei CM), deren Ergebnisse in Tabelle 3 dargestellt sind. Aufgrund der statistisch signifikanten Ergebnisse für die Endpunkte Reduktion der Migränetage pro Monat um ≥ 50 %, ≥ 75 % und 100 % erkannte der G-BA beträchtliche Vorteile zugunsten der Therapie mit Galcanezumab + BSC gegenüber Placebo + BSC, insbesondere bezüglich der Anzahl der Migränestunden pro Monat, der Reduktion der Kopfschmerztage pro Monat und der Veränderung des Migränezustands unter der Therapie. Bezüglich der Nebenwirkungen ergaben sich keine Unterschiede, es verblieben aber Unklarheiten bezüglich der Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext, weil es unklar war, inwiefern die ausgewerteten Patienten tatsächlich diejenigen Patienten repräsentieren, für die keine weiteren medikamentösen Therapien mehr infrage kommen und daher BSC als zweckmäßiger Vergleich angesehen werden kann. Daraus resultierte ein Anhaltspunkt für einen beträchtlichen Zusatznutzen (28).
Fremanezumab
Für die Nutzenbewertung von Fremanezumab wurden Daten vorgelegt aus den Zulassungsstudien HALO (CM) und HALO (EM) (siehe Tabelle 3) für Fragestellung 1 (unbehandelte Patienten oder solche, die auf mindestens eine prophylaktische Medikation nur unzureichend angesprochen haben) und Daten aus der Studie FOCUS für Fragestellung 3 (hochgradig therapierefraktäre Patienten). FOCUS war eine randomisierte, doppelblinde Studie mit einer zwölfwöchigen doppelblinden, placebokontrollierten Behandlungsphase und einer sich daran anschließenden zwölfwöchigen offenen Phase, in der alle Patienten Fremanezumab erhielten. In die Studie wurden insgesamt 838 Patienten mit seit mindestens zwölf Monaten dokumentierter CM oder EM eingeschlossen. Patienten mit EM mussten ≥ 6 und < 15 Kopfschmerztage und davon ≥ 4 Migränetage pro Monat innerhalb der Run-in-Phase aufweisen, Patienten mit CM ≥ 15 Kopfschmerztage und davon ≥ 8 Migränetage pro Monat. Zudem mussten die Patienten ein dokumentiertes Therapieversagen auf zwei bis vier der folgenden Wirtoksffe bzw. Wirkstoffklassen haben: Betablocker, Topiramat, Amitriptylin, Flunarizin, Candesartan, Clostridium botulinum Toxin Typ A und Valproinsäure. Die Patienten erhielten entweder 675 mg Fremanezumab als Anfangsdosis, gefolgt von 225 mg Fremanezumab einmal monatlich (n = 283); 675 mg Fremanezumab alle drei Monate und je eine Placebo-Injektion alle vier Wochen zwischen zwei Gaben von Fremanezumab (n = 276) oder monatliche Injektion von Placebo (n = 279) (29).
Der pharmazeutische Unternehmer bildete verschiedene Teilpopulationen aus diesen Studien für die Nutzenbewertung. Für die Zielpopulation „unbehandelte Patienten oder Patienten mit Therapieversagen gegenüber einer Therapie“ wurden als Vergleichsgruppe die Patienten des jeweiligen Placebo-Arms der HALO (CM)- und HALO (EM)-Studie selektiert, die während der Studie weiterhin ihre Begleitmedikationen zur Migräneprophylaxe einnahmen. Diese Daten wurden vom G-BA allerdings als nicht geeignet eingestuft, da im Vergleichsarm die Fortführung dieser bestehenden, aber unzureichenden Therapie nicht der Umsetzung der zweckmäßigen Vergleichstherapie entsprach. Der Zusatznutzen ist für diese Patientenpopulation nicht belegt.
Für Fragestellung 3 – hochgradig therapierefraktäre Patienten – wurde eine Teilpopulation aus der FOCUS-Studie selektiert, für die in der Vergangenheit die Einnahme von Valproinsäure dokumentiert war. Weil in Deutschland Valproinsäure gemäß Arzneimittel-Richtlinie grundsätzlich nur dann verordnet werden darf, wenn andere Arzneimittel nicht erfolgreich oder nicht geeignet sind, ging der pharmazeutische Unternehmer davon aus, dass für Patienten mit Valproinsäure-Therapie alle anderen Therapien bereits entweder ausprobiert wurden oder kontraindiziert sind. In dieser Patientenauswahl zeigten sich signifikante Vorteile für Fremanezumab bezüglich der Reduktion der Migränetage pro Monat um ≥ 50 % und ≥ 75 % sowie der Kopfschmerztage pro Monat. Auch in Bezug auf die gesundheitsbezogene Lebensqualität ergaben sich statistisch signifikante, klinisch relevante Vorteile für Fremanezumab gegenüber Placebo, bei den Nebenwirkungen ließen sich weder Vor- noch Nachteile ableiten. Daraus ergab sich für den G-BA ein Anhaltspunkt für einen beträchtlichen Zusatznutzen, es verblieben allerdings Unklarheiten bezüglich der Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext. Aus Sicht des G-BA kann nicht davon ausgegangen werden, dass in der deutschen Versorgungspraxis Patienten bereits nach zwei Vortherapien als therapieresistent oder insgesamt nicht mehr behandelbar gelten. Damit ist unklar, inwiefern die ausgewerteten Patienten tatsächlich diejenigen Patienten repräsentieren, für die keine weiteren medikamentösen Therapien mehr infrage kommen (30).
Fazit für die Praxis
In Deutschland sind bereits drei monoklonale Antikörper zur Prophylaxe von Migräne bei Erwachsenen mit mindestens vier Migränetagen pro Monat verfügbar. Die Wirkstoffe richten sich spezifisch gegen das migräneauslösende Neuropeptid CGRP und greifen damit einen für die Pathophysiologie der Migräne zentralen Mechanismus an. Für die Erstbehandlung und die meisten vorbehandelten Patienten liegen keine Daten aus direkt vergleichen klinischen Studien vor. Damit ist der eigentliche Stellenwert der CGRP-Antikörper noch unklar. Lediglich für die recht kleine Untergruppe der bislang therapieresistenten Patienten liegen Daten mäßiger Qualität vor und nur für diese Patienten wurde ein Zusatznutzen festgestellt. Der Einsatz von Erenumab, Galcanezumab und Fremanezumab sollte daher vorerst nur nach Versagen anderer Arzneimittel zur Migräneprophylaxe oder bei deren Unverträglichkeit erfolgen.
Interessenkonflikte
Ein Interessenkonflikt wird von den Autoren verneint.
Literatur
- GBD 2016 Headache Collaborators: Global, regional, and national burden of migraine and tension-type headache, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2018; 17: 954-976.
- Bigal ME, Lipton RB: The epidemiology, burden, and comorbidities of migraine. Neurol Clin 2009; 27: 321-334.
- Diener HC, Gaul C, Kropp P: Therapie der Migräneattacke und Prophylaxe der Migräne: www.dgn.org/leitlinien (letzter Zugriff: 6. September 2019). Leitlinien für Diagnostik und Therapie in der Neurologie. Deutsche Gesellschaft für Neurologie (Hrsg.), 2018.
- Acis Arzneimittel GmbH: Fachinformation "Flunarizin acis® 5 mg/10 mg Hartkapseln". Stand: Januar 2018.
- Janssen-Cilag GmbH: Fachinformation "Topiramat®-Janssen 25 mg/50 mg/100 mg/200 mg Filmtabletten". Stand: Juni 2019.
- Allergan Pharmaceuticals Ireland: Fachinformation "Botox® 50/100/200 Allergan-Einheiten Pulver zur Herstellung einer Injektionslösung". Stand: Januar 2019.
- Gemeinsamer Bundesausschuss (G-BA): Anlage VI zum Abschnitt K der Arzneimittel-Richtlinie: Verordnungsfähigkeit von zugelassenen Arzneimitteln in nicht zugelassenen Anwendungsgebieten (sog. Off-Label-Use): www.g-ba.de/downloads/83-691-497/AM-RL-VI-Off-label-2018-07-13.pdf (letzter Zugriff: 6. Februar 2019). Berlin, 13. Juli 2018.
- European Commission, Union Register of medicinal products: ec.europa.eu/health/documents/community-register/html/index_en.htm. Letzter Zugriff: 6. September 2019.
- Novartis Pharma GmbH: Fachinformation "Aimovig® 70 mg / 140 mg Injektionslösung in einer Fertigspritze / Injektionslösung im Fertigpen". Stand: August 2019.
- www.gkv-spitzenverband.de/krankenversicherung/arzneimittel/verhandlungen_nach_amnog/ebv_130b/ebv_nach_130b.jsp. Letzter Zugriff: 20. Oktober 2019.
- European Medicines Agency (EMA): Aimovig® - Emicizumab: European Assessment Report (EPAR) (Assessment Report): www.ema.europa.eu/documents/assessment-report/aimovig-epar-public-assessment-report_en.pdf (letzter Zugriff: 1. November 2019). London, 31. Mai 2018.
- European Medicines Agency (EMA): Emgality® - Galcanezumab: European Assessment Report (EPAR): www.ema.europa.eu/documents/assessment-report/emgality-epar-public-assessment-report_en.pdf (letzter Zugriff: 1. November 2019). London, 20. September 2018.
- European Medicines Agency (EMA): Ajovy® - Fremanezumab: European Assessment Report (EPAR): www.ema.europa.eu/documents/assessment-report/ajovy-epar-public-assessment-report_en.pdf (letzter Zugriff: 1. November 2019). London, 31. Januar 2019.
- Eli Lilly Nederland B.V.: Fachinformation "Emgality® 120 mg Injektionslösung in Fertigpen". Stand: November 2018.
- Teva GmbH: Fachinformation "Ajovy® 225 mg Injektionslösung in Fertigspritze". Stand: März 2019.
- Majima M, Ito Y, Hosono K, Amano H: CGRP/CGRP receptor antibodies: potential adverse effects due to blockade of neovascularization? Trends Pharmacol Sci 2019; 40: 11-21.
- Yallampalli C, Chauhan M, Endsley J, Sathishkumar K: Calcitonin gene related family peptides: importance in normal placental and fetal development. Adv Exp Med Biol 2014; 814: 229-240.
- Gangula PR, Thota C, Wimalawansa SJ et al.: Mechanisms involved in calcitonin gene-related Peptide-induced relaxation in pregnant rat uterine artery. Biol Reprod 2003; 69: 1635-1641.
- Tepper S, Ashina M, Reuter U et al.: Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 2017; 16: 425-434.
- Detke HC, Goadsby PJ, Wang S et al.: Galcanezumab in chronic migraine: The randomized, double-blind, placebo-controlled REGAIN study. Neurology 2018; 91: e2211-e2221.
- Silberstein SD, Dodick DW, Bigal ME et al.: Fremanezumab for the Preventive Treatment of Chronic Migraine. N Engl J Med 2017; 377: 2113-2122.
- Goadsby PJ, Reuter U, Hallstrom Y et al.: A controlled trial of erenumab for episodic migraine. N Engl J Med 2017; 377: 2123-2132.
- Stauffer VL, Dodick DW, Zhang Q et al.: Evaluation of galcanezumab for the prevention of episodic migraine: the EVOLVE-1 randomized clinical trial. JAMA Neurol 2018; 75: 1080-1088.
- Skljarevski V, Oakes TM, Zhang Q et al.: Effect of different doses of galcanezumab vs placebo for episodic migraine prevention: a randomized clinical trial. JAMA Neurol 2018; 75: 187-193.
- Dodick DW, Silberstein SD, Bigal ME et al.: Effect of fremanezumab compared with placebo for prevention of episodic migraine: a randomized clinical trial. JAMA 2018; 319: 1999-2008.
- Reuter U, Goadsby PJ, Lanteri-Minet M et al.: Efficacy and tolerability of erenumab in patients with episodic migraine in whom two-to-four previous preventive treatments were unsuccessful: a randomised, double-blind, placebo-controlled, phase 3b study. Lancet 2018; 392: 2280-2287.
- Gemeinsamer Bundesausschuss: Tragende Gründe zum Beschluss des Gemeinsamen Bundesausschusses über eine Änderung der Arzneimittel-Richtlinie (AM-RL): Anlage XII – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V – Erenumab: www.g-ba.de/downloads/40-268-5716/2019-05-02_AM-RL-XII_Erenumab_D-407_TrG.pdf (letzter Zugriff: 1. November 2019). Berlin, 2. Mai 2019.
- Gemeinsamer Bundesausschuss: Tragende Gründe zum Beschluss des Gemeinsamen Bundesausschusses über eine Änderung der Arzneimittel-Richtlinie (AM-RL): Anlage XII – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V – Galcanezumab: www.g-ba.de/downloads/40-268-6010/2019-09-19_AM-RL-XII_Galcanezumab_D-445_TrG.pdf (letzter Zugriff: 1. November 2019). Berlin, 19. September 2019.
- Ferrari MD, Diener HC, Ning X et al.: Fremanezumab versus placebo for migraine prevention in patients with documented failure to up to four migraine preventive medication classes (FOCUS): a randomised, double-blind, placebo-controlled, phase 3b trial. Lancet 2019; 394: 1030-1040.
- Gemeinsamer Bundesausschuss: Tragende Gründe zum Beschluss des Gemeinsamen Bundesausschusses über eine Änderung der Arzneimittel-Richtlinie (AM-RL): Anlage XII – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V – Fremanezumab: www.g-ba.de/downloads/40-268-6093/2019-11-07_AM-RL-XII_Fremanezumab_D-460_TrG.pdf (letzter Zugriff: 1. November 2019). Berlin, 7. November 2019.
vorab online
Dieser Artikel wurde am 17. Dezember 2019 vorab online veröffentlicht.