Europäische Fälschungsschutzrichtlinie und ihre Umsetzung in Deutschland
Falsified Medicines Directive und its implementation in Germany
Zusammenfassung
Die Fälschungsschutzrichtlinie (2011/62/EU) sieht ab Februar 2019 für viele Arzneimittel in der Europäischen Union obligatorische Sicherheitsmerkmale – ein individuelles Erkennungsmerkmal und eine Vorrichtung gegen Manipulation – auf der Umverpackung vor. Die End-to-End-Verifikation ermöglicht die Echtheitsprüfung mit dem Ziel, Patienten besser vor gefälschten Arzneimitteln in der legalen Vertriebskette zu schützen. In diesem Artikel wird die Umsetzung in Deutschland vorgestellt.
Abstract
The Falsified Medicines Directive (2011/62/EU) is to require obligatory safety features – a unique identifier and an anti-tampering device – on the outer packaging of many medicines in the European Union from February 2019. The end-to-end verification will guarantee medicine authenticity in order to protect patients from falsified medicines in the legal distribution chain. This article describes the implementation in Germany.
Rechtlicher Rahmen
Im Juli 2011 verabschiedete die Europäische Kommission (EC) die sogenannte Fälschungsschutzrichtlinie (2011/62/EU vom 8. Juni 2011; Falsified Medicines Directive) mit dem Ziel, Patienten in Europa besser vor gefälschten Arzneimitteln in der legalen Vertriebskette zu schützen. Anlass dafür waren etliche Fälle gefälschter Arzneimittel, die trotz der vorhandenen strengen Kontrollen immer wieder in der legalen Vertriebskette aufgedeckt worden waren (1). Die Richtlinie sieht die Einführung harmonisierter europäischer Maßnahmen zur Bekämpfung von Arzneimittelfälschungen vor, zu denen gehören u. a.:
- obligatorische Sicherheitsmerkmale auf der Umverpackung von Arzneimitteln
- ein gemeinsames, EU-weites Logo zur Kennzeichnung legaler Versand- bzw. Internetapotheken
- strengere Vorschriften für die Einfuhr von Arzneimittelwirkstoffen
- strengere Anforderungen an die Dokumentation für Großhändler.
Die technischen und organisatorischen Vorgaben zur Umsetzung der Fälschungsschutzrichtlinie wurden in der delegierten Verordnung (EU) Nr. 2016/161 konkretisiert (2). Damit sind sie aufgrund des Unionsrechts für alle Mitgliedstaaten automatisch und ohne nationale Umsetzung verbindliche, allgemeingeltende Regeln.
Vorgaben der delegierten Verordnung
Die wichtigste Maßnahme ist die „Echtheitsprüfung“ von Arzneimitteln in der legalen Vertriebskette, die anhand von zwei Sicherheitsmerkmalen erfolgen soll:
- ein individuelles Erkennungsmerkmal („unique identifier“), das die Überprüfung der Echtheit und die Identifizierung einer einzelnen Arzneimittelpackung ermöglicht
- eine Vorrichtung gegen Manipulation („anti-tampering device“, Erstöffnungsschutz, Originalitätsverschluss), mit der erkennbar ist, ob die äußere Verpackung eines Arzneimittels bereits geöffnet bzw. manipuliert wurde.
Das individuelle Erkennungsmerkmal beinhaltet vier Datenelemente: ein Produktcode, eine Seriennummer, die Chargenbezeichnung und das Verfalldatum (Abbildung 1). Im Produktcode ist in Deutschland u. a. die Pharmazentralnummer (PZN) des Arzneimittels enthalten, die eine eindeutige Identifikation von Fertigarzneimittelnamen, Darreichungsform, Wirkstärke, Packungsgröße und Verpackungsart ermöglicht. In anderen europäischen Ländern kann der Produktcode z. B. eine Kostenerstattungsnummer enthalten.
Die Seriennummer besteht aus einer maximal 20-stelligen Folge numerischer oder alphanumerischer Zeichen, die zufällig erstellt wird. Als Datenträger für das individuelle Erkennungsmerkmal fungiert ein zweidimensionaler Data Matrix Code, der die maschinelle Lesung (Abscannen) der Daten erlaubt.

Die Vorrichtung gegen Manipulation muss von jedem pharmazeutischen Unternehmer (pU) selbst umgesetzt werden. Das können z. B. mit Klebepunkten, Klebesiegeln oder perforierten Öffnungslaschen versiegelte Packungen sein, die eine Überprüfung ermöglichen, ob die Faltschachtel unversehrt ist.
Welche Arzneimittel sind betroffen?
Mit Sicherheitsmerkmalen zu versehen sind alle Humanarzneimittel, die verschreibungspflichtig und nicht als Ausnahme in der „White list“ (Anhang I der delegierten Verordnung (EU) 2016/161) aufgeführt sind. Zu den Ausnahmen gehören aktuell u. a.: homöopathische Arzneimittel, Radionuklide, medizinische Gase, Arzneimittel für neuartige Therapien, Allergenextrakte, Kontrastmittel und Lösungen für die parenterale Ernährung. Alle anderen verschreibungspflichtigen Arzneimittel müssen die Sicherheitsmerkmale tragen und bei der Abgabe an Patienten verifiziert werden.
Einige wenige nicht verschreibungspflichtige Arzneimittel sind in der „Black List“ (Anhang II der delegierten Verordnung (EU) 2016/161) benannt und damit zusätzlich von der Verifizierungspflicht umfasst. Aktuell sind das nur Arzneimittel mit dem Wirkstoff Omeprazol in den Wirkstärken 20 mg und 40 mg. Andere nicht verschreibungspflichtige Arzneimittel dürfen die Sicherheitsmerkmale nicht tragen, sie können aber können zum Schutz der Patienten mit Vorrichtungen zum Erkennen einer möglichen Manipulation versehen werden (3).
Umsetzung in der Praxis: End-to-End-Verifizierung
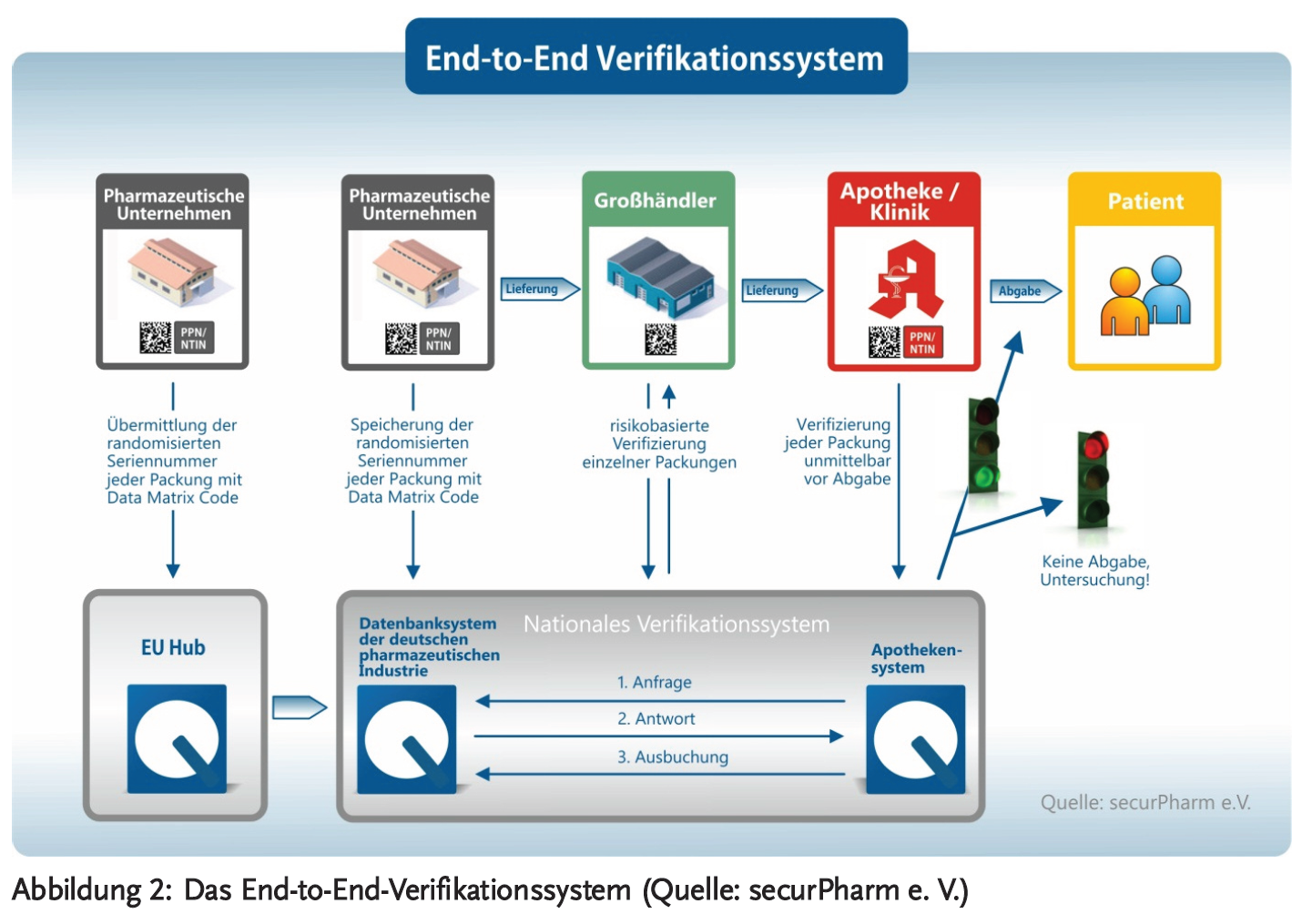
Ab dem 9. Februar 2019 dürfen pU verifizierungspflichtige Arzneimittel nur noch dann für den Verkehr freigeben, wenn diese Arzneimittelpackungen die zwei erforderlichen Sicherheitsmerkmale tragen. Beim Verpacken der Arzneimittel werden die Verkaufspackungen durch den pU mit den Sicherheitsmerkmalen versehen. Dabei wird für jede Packung eine individuelle Seriennummer generiert, die zusammen mit der PZN, Chargenbezeichnung und dem Verfalldatum in klarer Schrift und als Data Matrix Code auf die Packung aufgedruckt wird. Diese Daten werden vom pU in ein europaweites Datenbanksystem der pharmazeutischen Industrie hochgeladen.
Die Verifikation des Arzneimittels – also die Überprüfung, ob es ein echtes oder gefälschtes Medikament ist – erfolgt durch Abscannen vom Data Matrix Code und den damit ausgelösten Abgleich mit der Datenbank der pharmazeutischen Industrie. Dieser Schritt soll in der Apotheke vor der Abgabe an einen Patienten erfolgen. Die Rückmeldung gibt an, ob die zu verifizierende Kombination der Sicherheitsmerkmale tatsächlich existiert und wenn ja, wie der dort vermerkte Status der Packung ist. Bei Abgabe in der Apotheke wird die jeweilige Packung als entsprechend „abgegeben“ in der Datenbank eingetragen, also ausgebucht, die Seriennummer ist damit verbraucht. Sollte nochmal eine Packung mit der gleichen Nummer abgefragt werden, so würde die Software dies erkennen und das Arzneimittel als Fälschung melden. Die Verifikationsanfragen aus den Apotheken werden dabei anonymisiert über einen separaten Apothekenserver bearbeitet und weitergeleitet, damit der Datenschutz gewährleistet wird (Abbildung 2).
Falls die Rückmeldung erfolgt, dass die Verifizierung des Arzneimittels fehlgeschlagen ist, muss von einem möglichen Fälschungsverdacht ausgegangen werden. Die betroffene Packung muss separiert und darf nicht abgegeben werden.
Umsetzung in Deutschland
In Deutschland haben sich die Verbände der pharmazeutischen Unternehmen, des Großhandels und der Apothekerschaft zu der nicht gewinnorientierten Stakeholder-Organisation securPharm e. V. zusammengeschlossen, um die Umsetzung der Fälschungsschutzrichtlinie technisch und organisatorisch zu begleiten und das System zur Echtheitsprüfung von Arzneimitteln aufzubauen (4).
Folgen für die ärztliche Praxis
Ärzte sind primär nicht von der Umsetzung der Fälschungsschutzrichtlinie in Deutschland betroffen. Ärztemuster müssen vom pU vor der Abgabe an den Arzt aus dem System ausgebucht werden.
Arzneimittelpackungen, die vor dem Stichtag 9. Februar 2019 für das Inverkehrbringen freigegeben wurden, dürfen bis zum Ende ihrer Verfalllaufzeit abgegeben werden, unabhängig davon, ob sie die Sicherheitsmerkmale tragen oder nicht. Dies könnte dazu führen, dass ein und dasselbe Arzneimittel in drei verschiedenen Ausprägungen gleichzeitig im Markt verfügbar ist:
- ohne Sicherheitsmerkmale und ohne Vorrichtung gegen Manipulation
- mit Sicherheitsmerkmalen, aber bereits vor dem Stichtag in den Verkehr gebracht und daher nicht in der Datenbank der pharmazeutischen Industrie
- mit Sicherheitsmerkmalen und in der Datenbank der pharmazeutischen Industrie.
Das kann für Patienten verwirrend sein, sodass sie möglicherweise Rat bei ihrem Arzt suchen. Es ist auch zu erwarten, dass sich bei einigen Arzneimittel die „Aufmachung“ ändert: z. B. veränderte Größe und Form der Faltschachtel oder verändertes Design der Faltschachtel. Auch dies könnte zu Unsicherheit bei den Patienten führen.
Des Weiteren sind – insbesondere in der Anfangszeit – technische Probleme und Störungen nicht auszuschließen, die sich negativ auf die Arzneimittelversorgung auswirken könnten. Wenn z. B. aufgrund einer technischen Störung die Daten eines pU nicht in die Datenbank hochgeladen wurden oder nicht in der Datenbank freigeschalten wurden, dann könnten Arzneimittel bei der Abgabe in der Apotheke irrtümlich als Fälschungen eingestuft werden, die nicht abgegeben werden dürfen. Auch könnten Handhabungsfehler in der Apotheke (z.B. doppeltes Abscannen) einen Fehlalarm auslösen. Bei einem nicht so häufig verordneten Arzneimittel, das nicht in großer Menge vorrätig gehalten wird, könnte eine solche Störung (theoretisch) einen kurzfristigen Versorgungsengpass bedingen. Auch in solchen Fällen könnten sich verunsicherte Patienten an ihre Ärzte wenden.
Arzneimittelkommission der deutschen Ärzteschaft
Literatur
- Europäische Kommission: Falsified medicines: ec.europa.eu/health/human-use/falsified_medicines_de. Letzter Zugriff: 26. September 2018.
- Delegierte Verordnung (EU) 2016/161 der Kommission vom 2. Oktober 2015 zur Ergänzung der Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates durch die Festlegung genauer Bestimmungen über die Sicherheitsmerkmale auf der Verpackung von Humanarzneimitteln: eur-lex.europa.eu/legal-content/DE/TXT/. Letzter Zugriff: 26. September 2018.
- Bundesinstitut für Arzneimittel und Medizinprodukte und Paul-Ehrlich-Institut Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel: Gemeinsame Bekanntmachung zur Umsetzung des Artikels 54a Absatz 5 Satz 3 der Richtlinie 2001/83/EG zur Vorrichtung gegen Manipulationen (anti-tampering device). Stand: 11. April 2017. Veröffentlicht am 26. April 2017, BAnz AT 26.04.2017 B3.
- www.securpharm.de. Letzter Zugriff: 26. September 2018.
Weitere Informationen
www.pei.de/SharedDocs/Downloads/vigilanz/bulletin-zur-arzneimittelsicherheit/2018/3-2018.pdf
vorab online
Dieser Artikel wurde am 14. November 2018 vorab online veröffentlicht.