Emicizumab (Hemlibra®) ▼
Zugelassene Indikation und Wirkmechanismus
Emicizumab (Hemlibra®) ist zur subkutanen Gabe als Routineprophylaxe von Blutungsereignissen bei Patienten mit Hämophilie A und Faktor-VIII-Hemmkörpern in allen Altersgruppen zugelassen. Emicizumab ist ein chimärer, bispezifischer, humanisierter monoklonaler Antikörper, der den aktivierten Gerinnungsfaktor IX und den Gerinnungsfaktor X verbindet, um die Funktion vom fehlenden aktivierten Faktor VIII nachzuahmen.
Bewertung
Hemlibra® (Emicizumab) wurde im Februar 2018 anhand der Daten aus den HAVEN-1- und HAVEN-2-Studien (Phase III) zugelassen. Diese vorläufigen Daten zeigen eine signifikante Reduktion der behandlungsbedürftigen Blutungen durch die prophylaktische wöchentliche Gabe von Emicizumab bei Jugendlichen und Erwachsenen mit Hämophilie A und Faktor-VIII-Hemmkörpern. Auch wurde dadurch eine signifikante Verbesserung der gesundheitsbezogenen Lebensqualität und des Gesundheitszustandes erzielt. Auch Kinder unter 12 Jahren scheinen von der Prophylaxe mit Emicizumab zu profitieren. In der Studie HAVEN 2 hatten 87 % unter Emicizumab keine behandlungsbedürftige Blutung und 34,8 % hatten keine Blutung. Emicizimab erscheint damit als eine subkutane Alternative zur intravenösen Therapie mit Faktor-VIII-Konzentraten und FVIII-Bypassing-Präparaten. Es fehlen allerdings Langzeitdaten zur Wirksamkeit, Sicherheit und Immunogenität von Emicizumab, sodass eine abschließende Bewertung des Nutzen-Risiko-Profils nicht möglich ist. Auch ist der Metabolismus von Emicizumab nicht vollständig untersucht worden. Dies ist aber für einen IgG4-basierten Antikörper zwingend erforderlich, da IgG4-vermittelte Autoimmunerkrankungen mit variabler klinischer Präsentation wie beispielsweise Autoimmunpankreatitis, IgG4-assozierte Hepatitis und Cholangiopathie und Mikulicz-Syndrom bekannt sind.
Markteinführung
Hemlibra® (Emicizumab) ist seit dem 01.04.2018 auf dem deutschen Arzneimittelmarkt verfügbar.
Wirksamkeit in den Zulassungsstudien
Die beiden Hauptzulassungsstudien HAVEN 1 und HAVEN 2 sind noch nicht abgeschlossen. Die Zulassung basiert auf den vorläufigen Ergebnissen. Hemlibra® wurde durch eine beschleunigte Beurteilung („accelerated assessment“) im Rahmen des zentralisierten Zulassungsverfahrens in der EU zugelassen. Eine solche Beurteilung ist möglich für Arzneimittel, die von besonderer Bedeutung für die öffentliche Gesundheit sind, insbesondere für diejenigen, die therapeutische Innovationen darstellen.
Die Studie HAVEN 1 (NCT02622321, Sponsor Hoffmann-La Roche) ist eine multizentrische, offene, randomisierte, kontrollierte Phase-III-Studie an 109 männlichen Jugendlichen und Erwachsenen (im Alter von 12 bis 75 Jahren) mit Hämophilie A und Faktor-VIII-Hemmkörpern (mit hohem Titer: ≥ 5 Bethesda-Einheiten (BE)). Die Studienteilnehmer waren zuvor entweder episodisch („on demand“) oder prophylaktisch mit FVIII-Bypassing-Präparaten (aktiviertes Prothrombinkomplex-Konzentrat (aPCC)/factor-eight-inhibitor bypass activity (FEIBA) und rekombinanter Faktor-VIIa (rFVIIa)) behandelt worden. Die 53 on-demand (bei Bedarf) risikoadjustiert behandelten Patienten wurden stratifiziert nach der Blutungsrate in den vorangegangenen 24 Wochen (< 9 oder ≥ 9) im Verhältnis 2 : 1 randomisiert und erhielten entweder Emicizumab (Arm A) oder keine Prophylaxe (Arm B). Die zuvor prophylaktisch-kontinuierlich behandelten 49 Patienten wurden dem Arm C zugeteilt und erhielten Emicizumab. Primärer Endpunkt war die Wirksamkeit von Emicizumab innerhalb von 24 Wochen im Vergleich zu keiner Prophylaxe (Arm A vs. Arm B) evaluiert nach Anzahl der Blutungsereignisse, die mit Gerinnungsfaktoren behandelt werden mussten. Blutungen aufgrund von Operationen und Behandlungen wurden nicht als Ereignis gewertet. Sekundäre Endpunkte waren die Anzahl aller Blutungen, spontaner Blutungen, der Gelenkblutungen und der Blutungen in Zielgelenken sowie die gesundheitsbezogene Lebensqualität und der Gesundheitszustand der Patienten. Des Weiteren wurde die Wirksamkeit von Emicizumab im Vergleich zwischen zuvor on demand und prophylaktisch behandelten Patienten (Arm A versus Arm C) untersucht. Etwa die Hälfte der Patienten im Arm C war vor Aufnahme in die HAVEN-1-Studie in einer nichtinterventionellen Studie (NIS) eingeschlossen. Die dabei erfasste Prophylaxe mit Bypassing-Präparaten wurde mit der Prophylaxe mit Emicizumab verglichen (Tabelle 2).
In den Tabellen 1, 2 und 3 sind die Ergebnisse für die patientenrelevanten Endpunkte dargestellt.
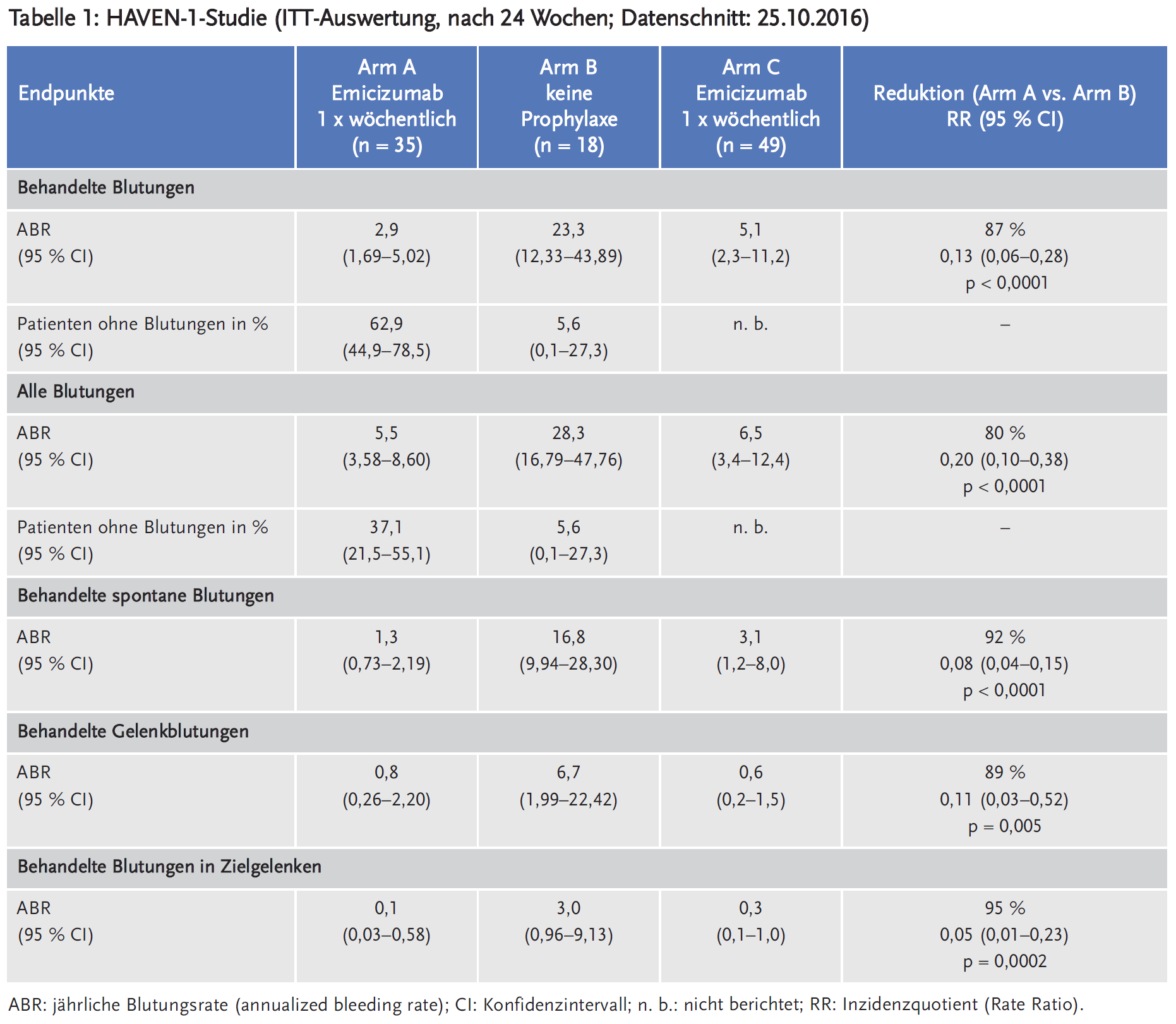
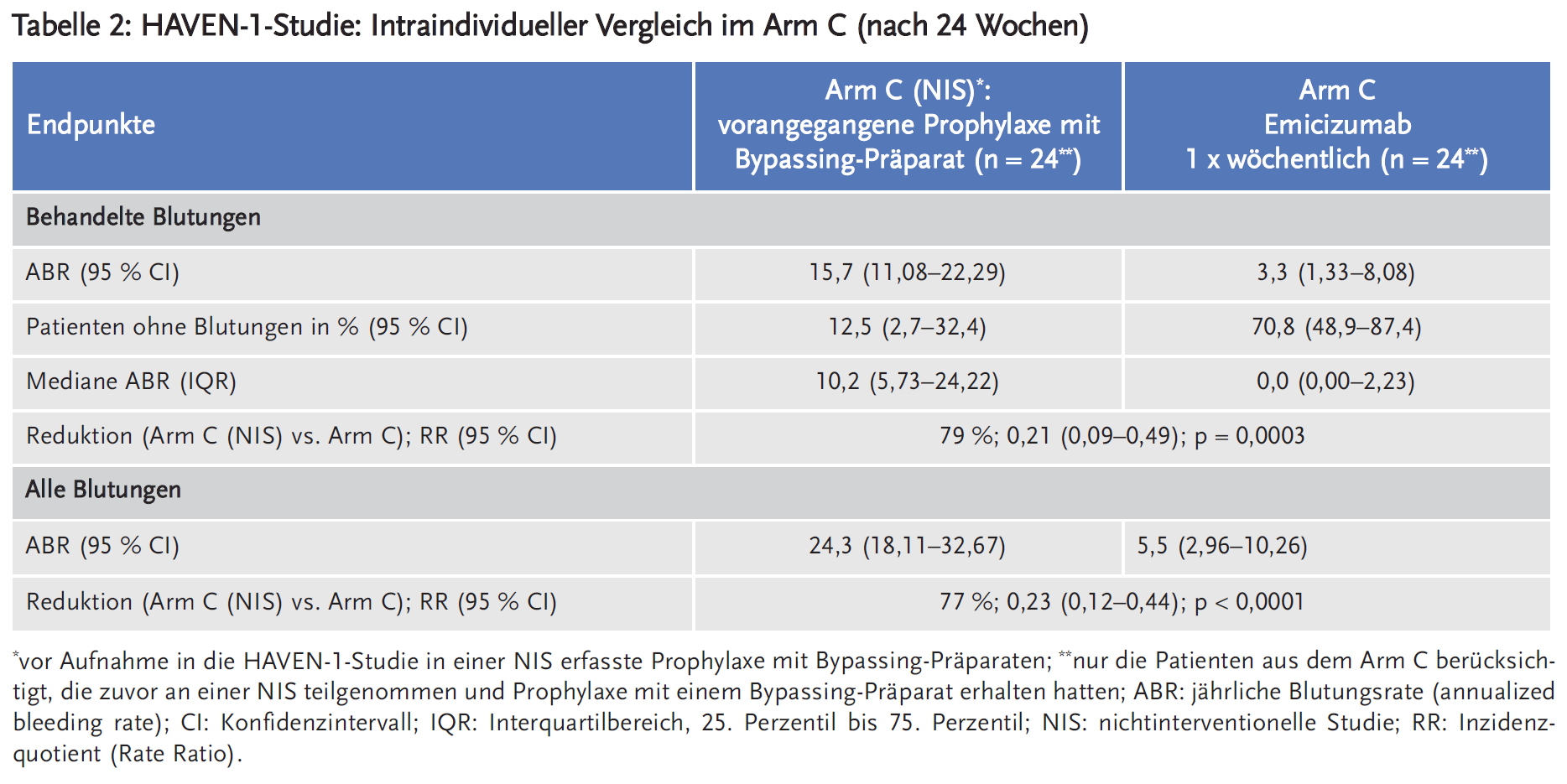
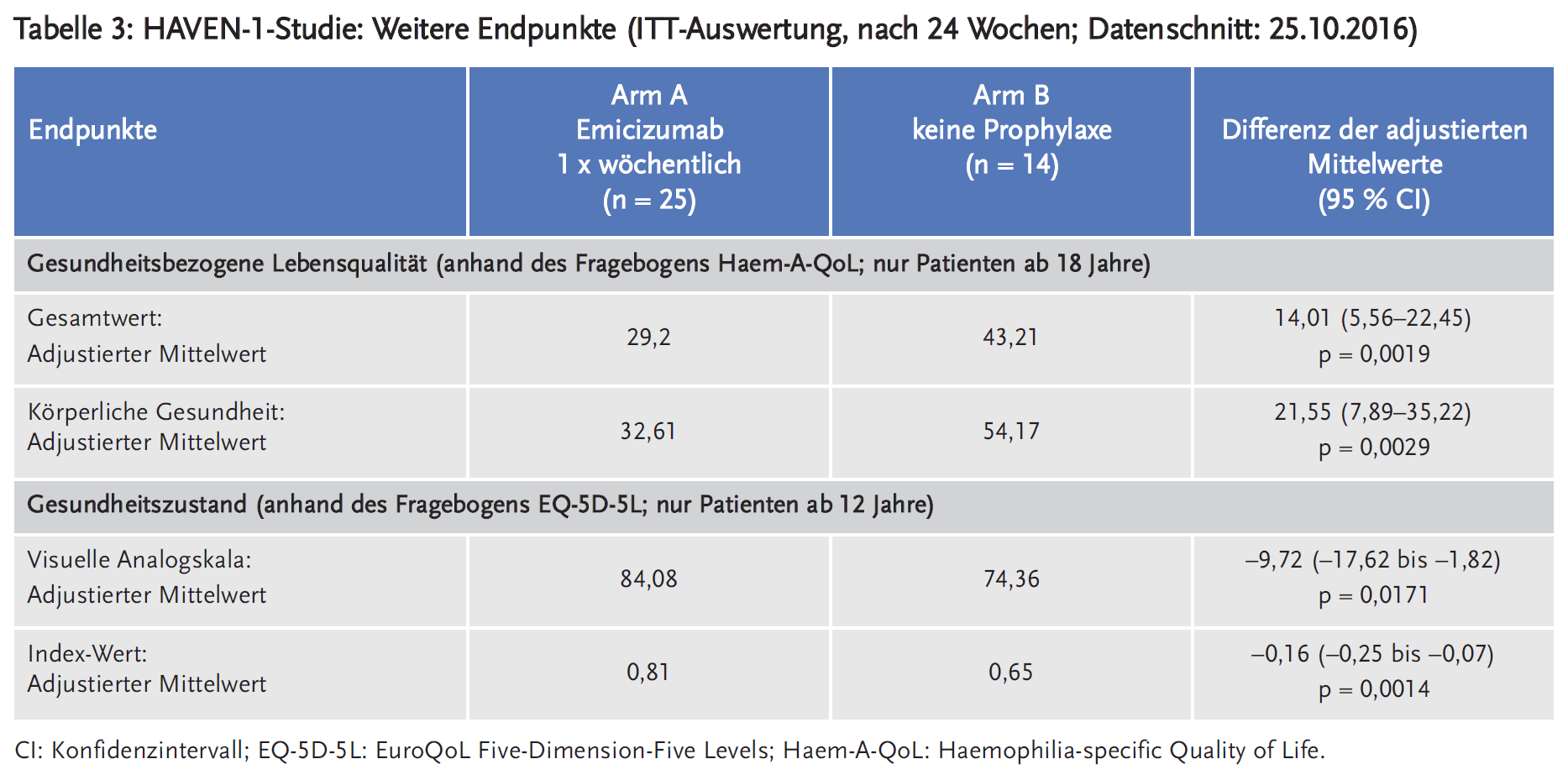
Bei keinem Patienten war der Test auf Antikörper gegen Emicizumab (Antidrug-Antikörper, ADA) positiv, jedoch zeigten zwei Patienten im Behandlungsverlauf Veränderungen der pharmakokinetischen Parameter, die indirekt auf ADA hinwiesen. Während der 24-wöchigen Behandlung mit Emicizumab blieben die Titer der Faktor-VIII-Hemmkörper bei den meisten Patienten stabil oder zeigten einen Abfall.
Die Studie HAVEN 2 (NCT02795767; Sponsor Hoffmann-La Roche) ist eine offene, multizentrische, einarmige Phase-III-Studie, in die Kinder (unter 12 Jahren) und Jugendlichen (im Alter von 12 bis 17 Jahren mit einem Körpergewicht unter 40 kg) mit Hämophilie A und Faktor-VIII-Hemmkörpern eingeschlossen werden. Für die Zulassung reichte der pU die Daten einer Interimsanalyse (Datenschnitt: 28.10.2016) ein. Zu diesem Zeitpunkt waren 23 Patienten eingeschlossen, die zuvor entweder episodisch oder prophylaktisch mit Bypassing-Präparaten behandelt worden waren. Primärer Endpunkt war die Wirksamkeit von Emicizumab, evaluiert nach Anzahl der Blutungen, die mit Gerinnungsfaktoren behandelt werden mussten. Sekundäre Endpunkte waren die Anzahl aller Blutungen, spontaner Blutungen, der Gelenkblutungen und der Blutungen in Zielgelenken.
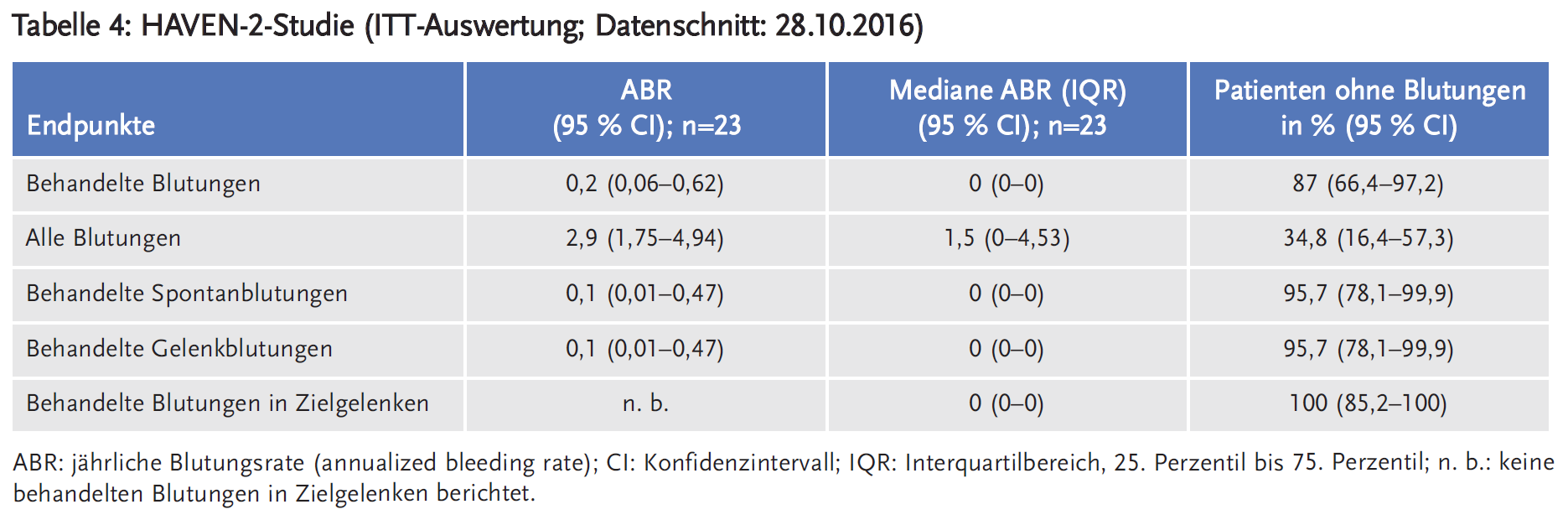
Basierend auf diesen vorläufigen Daten zeigt sich eine statistisch signifikante Reduktion der Häufigkeit behandlungsbedürftiger Blutungen sowie aller Blutungen bei Patienten mit Hämophilie A und Faktor-VIII-Hemmkörpern durch die prophylaktische wöchentliche Gabe von Emicizumab. Auch wurde dadurch eine signifikante Verbesserung der gesundheitsbezogenen Lebensqualität und des Gesundheitszustandes erzielt. Auch Kinder unter 12 Jahren scheinen von der Anwendung von Emicizumab zu profitieren, 87 % der Kinder hatten in der Studie HAVEN 2 keine behandlungsbedürftige Blutung, 34,8 % keine Blutung. Die Vermeidung von Gelenkblutungen ist eines der wesentlichen Therapieziele bei Hämophilie A, da ein Haemarthros dauerhafte Schäden und Bewegungseinschränkungen verursacht. Der Verlauf des Gelenkfunktions-Score „mHJHs“ (modified Haemophilia Joint Health Score) unter der Prophylaxetherapie sollte im Rahmen einer Produktbeobachtung erfolgen.
Ausgewählte Nebenwirkungen
Die wichtigsten häufigen Nebenwirkungen waren Reaktionen an der Injektionsstelle, Kopfschmerzen, Fieber, Durchfall, Myalgie und Arthralgie. Als schwerwiegende Nebenwirkungen traten thrombotische Mikroangiopathien und thrombotische Ereignisse einschließlich Thrombosen des Sinus cavernosus auf, sowie oberflächliche Thrombophlebitiden begleitet von Hautnekrosen.
Ausgewählte Warnhinweise
Emicizumab ist in allen Altersgruppen zugelassen. Für Patienten im Alter von < 1 Jahr sowie >75 Jahren liegen keine Daten aus klinischen Studien vor.
Rückverfolgbarkeit: Um die Rückverfolgbarkeit von biologischen Arzneimitteln zu verbessern, sollten der Handelsname und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Thrombotische Mikroangiopathie und Thromboembolie: In klinischen Studien wurden Fälle thrombotischer Mikroangiopathie (TMA) sowie Thromboembolien beobachtet, die bei gleichzeitiger Gabe von Emicizumab und aPCC/FEIBA auftraten. Die gleichzeitige Anwendung von Emicizumab und aPCC/FEIBA sollte deshalb vermieden werden. Wenn Patienten unter Prophylaxe mit Emicizumab zusätzlich aPCC/FEIBA erhalten, sollten sie auf die Entwicklung einer Thromboembolie hin überwacht werden. Bei klinischen Symptomen, bildgebenden Untersuchungsbefunden und/oder Laborergebnissen, die auf thrombotische Ereignisse hindeuten, muss die Gabe von aPCC/FEIBA abgebrochen und die Therapie mit Emicizumab unterbrochen werden.
Gleichzeitige Gabe von Bypassing-Präparaten: Emicizumab erhöht das Gerinnungspotenzial, sodass die erforderliche Dosis des Bypassing-Präparates niedriger ist als ohne Prophylaxe mit Emicizumab. Die Dosis und die Anwendungsdauer des Bypassing-Präparates hängen von der Lokalisation und dem Ausmaß der Blutung sowie dem klinischen Zustand des Patienten ab. Die Anwendung von aPCC sollte vermieden werden, es sei denn, es stehen keine anderen Therapieoptionen/-alternativen zur Verfügung.
Einfluss auf Gerinnungstests: Emicizumab ersetzt die Kofaktor-Aktivität des aktivierten Faktor VIII (FVIIIa) im Tenase-Komplex. Gerinnungstests, die auf intrinsischer Gerinnung basieren, einschließlich aktivierter Gerinnungszeit (ACT) und aktivierter partieller Thromboplastinzeit (aPTT), messen die Gesamtgerinnungszeit einschließlich der Zeit, die für die Aktivierung von FVIII zu FVIIIa durch Thrombin benötigt wird. Mit solchen Tests wird unter Emicizumab eine übermäßig verkürzte Gerinnungszeit gemessen, die alle auf aPTT basierenden Einzelfaktor-Assays wie den Einstufentest der FVIII-Aktivität verfälscht. Einzelfaktor-Assays, die chromogene Peptidsubstrate oder immunbasierte Methoden anwenden, werden hingegen nicht beeinflusst und können zur Überwachung der Gerinnungsparameter während der Behandlung eingesetzt werden. Eine Bestimmung der Bethesda-Einheiten ist mit einem chromogenen FVIII-Assay möglich, der Gerinnungsfaktoren tierischen Ursprungs einsetzt, die von Emicizumab nicht erkannt werden.
Schulungsmaterial
Für einzelne Arzneimittel wird bereits bei der Zulassung angeordnet, dass das Arzneimittel nur unter Verwendung von Schulungsmaterialien in Verkehr gebracht werden darf. Das Schulungsmaterial dient dazu, die Wissensvermittlung zu optimieren und Hilfe bei der sicheren Anwendung des Arzneimittels zu geben, ggfs. unter Einbeziehung einer patientenbezogenen Ansprache. Das behördlich beauflagte und genehmigte Schulungsmaterial zu Hemlibra® ist verfügbar unter: https://www.pei.de/SharedDocs/Downloads/vigilanz/schulungsmaterial/Hemlibra-Schulungsmaterial-Aerzte_Version-2.0_Leitfaden-Fachpersonal.pdf?__blob=publicationFile&v=3.

Weiterführende Informationen
Das IQWiG wurde am 01.04.2018 mit der Bewertung des Zusatznutzens beauftragt, über den der G-BA entscheiden wird. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Hemlibra®, erschienen am 1. März 2018. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.).
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 28. Mai 2018 vorab online veröffentlicht.