Brodalumab (Kyntheum®) (frühe Nutzenbewertung)
In Kürze
- Mit dem Interleukin-Antagonisten Brodalumab wurde ein weiteres biologisch hergestelltes Arzneimittel für Patienten mit einer Plaque-Psoriasis zugelassen und in den Markt eingeführt.
- Für die Bewertung des Zusatznutzens von Brodalumab für die Behandlung von Erwachsenen mit mittelschwerer bis schwerer Plaque-Psoriasis, für die eine systemische Therapie infrage kommt, wurde vom pharmazeutischen Unternehmer (pU) keine direkt vergleichende Studie mit der vom G-BA festgelegten Vergleichstherapie vorgelegt. Der vom pU durchgeführte adjustierte indirekte Vergleich war aufgrund der jeweils zu kurzen Behandlungsdauer in den Studien für die Beantwortung der vorliegenden Fragestellung nicht geeignet. Ein Zusatznutzen war somit für das IQWiG nicht zu belegen.
Für Erwachsene, die auf andere systemische Therapien unzureichend angesprochen haben oder für die diese nicht infrage kommen, sieht das IQWiG positive Effekte von Brodalumab in den Endpunktkategorien Morbidität und gesundheitsbezogene Lebensqualität. Hinsichtlich der Morbidität besteht für den Endpunkt Remission (PASI 100) ein Hinweis auf einen Zusatznutzen und im Bereich der gesundheitsbezogenen Lebensqualität für den Endpunkt DLQI (0 oder 1) ein Anhaltspunkt für einen Zusatznutzen. In beiden Fällen ist das Ausmaß des Zusatznutzens nicht quantifizierbar, wobei es nach Auffassung des IQWiG für PASI 100 höchstens beträchtlich ist. - Die AkdÄ sieht für die Patientengruppe mit mittelschwerer bis schwerer Plaque-Psoriasis ebenso wie das IQWiG aufgrund der fehlenden bzw. nicht geeigneten Daten keinen Zusatznutzen für Brodalumab. Für Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die auf eine systemische Therapie inklusive Phototherapie nicht angesprochen oder diese nicht vertragen haben oder für diese nicht geeignet sind, sieht die AkdÄ einen Hinweis auf einen geringen Zusatznutzen gegenüber der zweckmäßigen Vergleichstherapie Ustekinumab.
- Bei Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die auf andere systemische Therapien nicht angesprochen haben, oder bei denen eine Kontraindikation oder Unverträglichkeit gegenüber solchen Therapien vorlag sieht der G-BA für Brodalumab positive Effekte bei der Morbidität (PASI 100) und der gesundheitsbezogenen Lebensqualität, die für den G-BA durch negative Effekte bei den nicht schweren Nebenwirkungen sowie beim Endpunkt allgemeine Erkrankungen und Beschwerden am Verabreichungsort nicht infrage gestellt wurden.
Die Plaque-Psoriasis ist mit einer Prävalenz von 2 % in der europäischstämmigen Bevölkerung eine häufige Hauterkrankung. Dem Krankheitsverlauf und den Leitlinien entsprechend erfolgt die Behandlung einer Plaque-Psoriasis primär mit topischen Wirkstoffen. Die systemische Therapie bleibt schweren, therapieresistenten Formen der Psoriasis vorbehalten (z. B. chronisch aktive, großflächige Psoriasis, psoriatische Erythrodermie).
- Für die Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische Therapie infrage kommen, sind grundsätzlich die Wirkstoffe Acitretin, Ciclosporin, Fumarsäureester und Methotrexat (MTX) zugelassen.
- Für die Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die auf andere systemische Therapien einschließlich Ciclosporin, MTX oder PUVA nicht ansprechen, diese nicht vertragen oder Kontraindikationen dagegen haben, sind grundsätzlich die TNF-alpha-Inhibitoren Adalimumab, Infliximab und Etanercept, die Interleukin-Antagonisten Guselkumab, Ixekizumab, Secukinumab und Ustekinumab sowie der PDE-Antagonisten Apremilast und der Wirkstoff Dimethylfumarat zugelassen.
Mit dem Interleukin-Antagonisten Brodalumab wurde ein weiteres biologisch hergestelltes Arzneimittel für diese Patientengruppen zugelassen und in den Markt eingeführt.
Für das Verfahren der frühen Nutzenbewertung nach § 35a SGB V ergaben sich für die Bewertung des Zusatznutzens von Brodalumab im Vergleich zur zweckmäßigen Vergleichstherapie (ZVT) bei erwachsenen Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische Therapie infrage kommen, zwei Fragestellungen (siehe Tabelle 1).
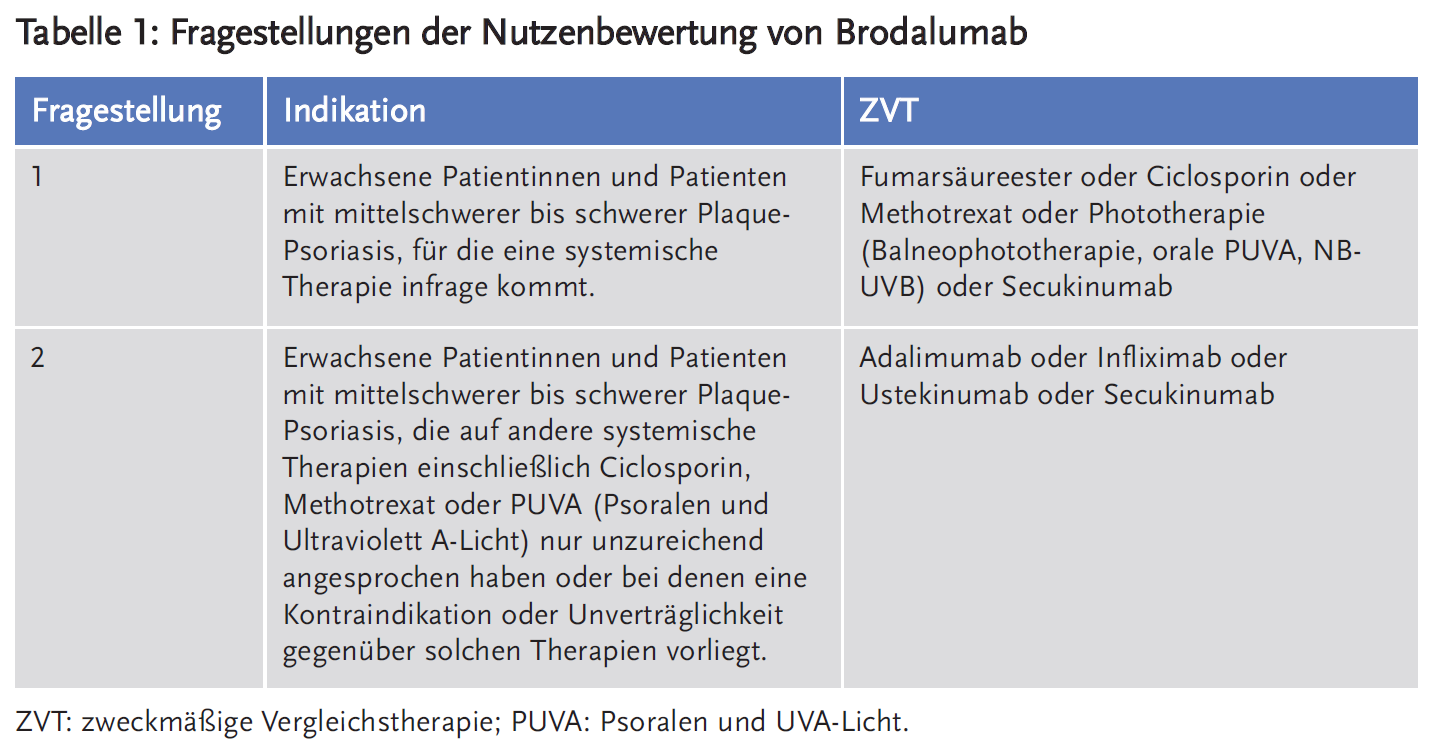
Fragestellung 1
- Für die Bewertung des Zusatznutzens von Brodalumab bei erwachsenen Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische und/oder Phototherapie geeignet sind, lagen weder für das IQWiG noch für die AkdÄ verwertbare Daten vor, sodass im Vergleich zur zweckmäßigen Vergleichstherapie kein Zusatznutzen gesehen wurde (1;2).
- Der pU hat keine Studie mit einem direkten Vergleich mit der zweckmäßigen Vergleichstherapie vorgelegt. Stattdessen legte er einen adjustierten indirekten Vergleich mit Brodalumab unter Verwendung der Studien AMAGINE-1, AMAGINE-2 und AMAGINE-3 (über die Dauer von 12 Wochen) und einen mit Fumarsäureester anhand der Studie BRIDGE (über die Dauer von 16 Wochen) vor. Die Vergleiche waren aufgrund der zu kurzen Behandlungsdauer für die Nutzenbewertung jedoch nicht geeignet (als notwendig erachtet wird für Therapien zur Behandlung der Plaque Psoriasis eine Mindeststudiendauer von 24 Wochen). Vor diesem Hintergrund kam der G-BA auf der Grundlage der Bewertung des IQWiG zu dem Ergebnis, dass ein Zusatznutzen von Brodalumab nicht belegt ist (3-5) (siehe Tabelle 2).

Fragestellung 2
Grundlage für die Bewertung der Fragestellung 2 durch das IQWiG waren die randomisierten, doppelblinden, multizentrischen Parallelgruppenstudien AMAGINE-2 und AMAGINE-3. Bei beiden Studien handelt es sich um mehrarmige Studien, in denen unterschiedliche Dosierungen von Brodalumab (Dosisfindung) mit Ustekinumab und mit Placebo verglichen wurden. Als relevante Teilpopulationen wurden nur der Arm mit der zugelassenen Dosierung von 210 mg Brodalumab alle zwei Wochen (15 % der ursprünglich eingeschlossenen Patienten) sowie der Ustekinumab-Arm (51 % der ursprünglich eingeschlossenen Patienten) ausgewertet. In der Metaanalyse beider Studien zeigten sich statistisch signifikante Unterschiede zugunsten von Brodalumab hinsichtlich der Morbiditätsendpunkte PASI 100, PASI 90, PASI 75, PSI sowie der gesundheitsbezogenen Lebensqualität. Bei den Nebenwirkungen zeigten sich weder Vor- noch Nachteile. Die vorgelegten Responderanalysen basierend auf dem PASI 100 werden aufgrund der insgesamt unklaren Datenlage als hoch verzerrt eingestuft, da der pU einen großen Anteil an Patienten, die auf die Therapie nicht angesprochen haben, als Non-Responder gewertet hat (31 % der Patienten im Brodalumab-Arm und 51 % der Patienten im Ustekinumab-Arm). Vor diesem Hintergrund wurden Sensitivitätsanalysen durchgeführt. Diese ergaben nur noch für PASI 100 und PASI 90 statistisch signifikante Ergebnisse zugunsten von Brodalumab. Der G-BA konstatierte daher unter Einbeziehung des dem IQWiG in Auftrag gegebenen Addendums einen Hinweis auf einen nicht quantifizierbaren Zusatznutzen gegenüber der ZVT Ustekinumab (2-4;6).
Für die AkdÄ liegen zur Fragestellung 2 zwar zwei gleich angelegte RCT mit weitgehend ähnlichen Ergebnissen vor; die Aussagesicherheit der für die Bewertung relevanten Patientenkollektive (180 für Brodalumab und 314 für Ustekinumab) ist jedoch begrenzt. Hinzu kommen die beschriebenen Unsicherheiten bez. Selektion der relevanten Patientenpopulation und bei der Bestimmung der Remissionsraten (Rettungsphase) sowie v. a. auch bei der Ermittlung der Verträglichkeit (Mangel an verlässlichen Daten). Bei Betrachtung des PASI 100 – für den Zusatznutzen am aussagekräftigsten – sind die kompletten Remissionsraten nach 52 Wochen im besten Fall 29,5 % (AMAGINE-2) bzw. 24,6 % (AMAGINE-3) höher als unter Ustekinumab und im schlechtesten Fall (Sensitivitätsanalysen) um 18,4 % bzw. 13,1 %. Dem steht eine möglicherweise schlechtere Verträglichkeit gegenüber. Das Ausmaß der möglicherweise schlechteren Verträglichkeit ist (unter Betrachtung aller vorliegenden Daten) aber eher als gering einzustufen. Wenn man von einer Verbesserung der Rate an kompletten Remissionen unter Berücksichtigung der Ergebnisse der Sensitivitätsanalysen zwischen 18 % und 13 % ausgeht – ohne Zunahme relevanter Nebenwirkungen – ist das Ausmaß des Zusatznutzen als gering einzustufen.
Die AkdÄ sieht daher für Brodalumab gegenüber der ZVT Ustekinumab einen Hinweis für einen geringen Zusatznutzen (1) (siehe Tabelle 3)

Literatur
- Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ): Stellungnahme der AkdÄ zur Nutzenbewertung nach § 35a SGB V - Brodalumab, Nr. 565, A17-42, Version 1.1, Stand: 1. Dezember 2017: www.akdae.de/Stellungnahmen/AMNOG/A-Z/Brodalumab/Brodalumab-EB.pdf (letzter Zugriff: 25. Mai 5.2018). Berlin, 22. Dezember 2017.
- Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG): IQWiG-Berichte - Nr. 565 Brodalumab (Plaque Psoriasis) - Nutzenbewertung gemäß § 35a SGB V - Auftrag A17-07 - Version 1.1: www.iqwig.de/download/A17-42_Brodalumab_Nutzenbewertung-35a-SGB-V_V1-1.pdf. Stand: 1. Dezember 2017.
- Gemeinsamer Bundesausschuss (G-BA): Tragende Gründe zum Beschluss des Gemeinsamen Bundesausschusses über eine Änderung der Arzneimittel-Richtlinie (AM-RL): Anlage XII - Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V - Brodalumab: www.g-ba.de/downloads/40-268-4854/2018-03-01_AM-RL-XII_Brodalumab_D-309_TrG.pdf. Berlin, 1. März 2018.
- Gemeinsamer Bundesausschuss (G-BA): Beschluss des Gemeinsamen Bundesausschusses über eine Änderung der Arzneimittel-Richtlinie (AM-RL): Anlage XII - Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V - Brodalumab: www.bundesanzeiger.de. Berlin, 22. März 2018.
- Kassenärztliche Bundesvereinigung (KBV): Frühe Nutzenbewertung - Brodalumab: www.kbv.de/html/33072.php. Letzter Zugriff: 25. Mai 2018.
- Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG): IQWiG-Berichte - Nr. 587 Brodalumab - Addendum zum Auftrag A17-42 (Ixekizumab, Plaque Psoriasis) - Nutzenbewertung gemäß § 35a SGB V - Auftrag A18-02 - Version 1.0: www.iqwig.de/download/A18-02_Brodalumab_Addendum-zum-Auftrag-A17-42_V1-0.pdf. Stand: 26. Januar 2018.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 27. Juni 2018 vorab online veröffentlicht.