Epidemiologie und Therapie von Infektionen durch Carbapenem-resistente Enterobakterien (CRE) in Deutschland
Zusammenfassung
Multiresistente, gramnegative Erreger (MRGN) gewinnen unter den ca. 400.000 bis 600.000 nosokomialen Infektionen, die jedes Jahr in Deutschland auftreten, zunehmend an Bedeutung. Ein besonderes Problem stellen dabei Carbapenemase-bildende Enterobacteriaceae dar, insbesondere Klebsiella pneumoniae. Entsprechende Stämme mit Produktion der wichtigsten bislang bekannten Carbapenemasen OXA-48, KPC, VIM, IMP und NDM-1 sind in Deutschland noch selten, machen im südeuropäischen (Reise-)Ausland (insbesondere Griechenland und Italien) in Hochrisikobereichen wie der Intensivmedizin jedoch bereits bis zu 70 % der klinischen Klebsiella-Isolate aus. Die genannten Resistenzmechanismen implizieren Kreuzresistenzen gegenüber einer Vielzahl anderer Antibiotika. Schwere Infektionen durch Carbapenem-resistente Enterobakterien (CRE) werden daher in der Regel mit Kombinationen sogenannter „Reserveantibiotika“ therapiert. Die vorgestellten Behandlungsprotokolle basieren fast ausschließlich auf retrospektiven und nicht randomisierten Studien. Auch die wenigen absehbaren Antibiotika-Neuentwicklungen bieten in dieser klinischen Situation nur einen eingeschränkten Fortschritt.
Abstract
Multiresistant, gram-negative bacteria rapidly gain importance among the approximately 400,000 to 600,000 nosocomial infections that occur annually in Germany. A particular problem is posed by carbapenemase producing Enterobacteriaceae, especially Klebsiella pneumoniae. Carbapenemases (mainly OXA-48, KPC, VIM, IMP, and NDM-1) producing strains are still rare in Germany, but are detectable in the countries of Southern Europe (particularly Greece and Italy) in high risk areas such as intensive care in up to 70 % of clinical Klebsiella isolates. The mechanisms of carbapenem-resistance implicate cross-resistance to a variety of other antibiotics. Severe infections with carbapenem-resistant Enterobacteriaceae (CRE) are usually treated with combinations of “reserve antibiotics”. The presented treatment protocols are almost exclusively based on retrospective, non-randomized studies. Furthermore, the few new developments of antibiotics offer only limited advancement in this clinical situation.
Einführung
Laut Daten des Krankenhaus-Infektions-Surveillance-Systems (KISS) (www.nrz-hygiene.de) sowie zweier umfassender nationaler Prävalenzstudien aus den Jahren 1994 und 2011 ereignen sich in Deutschland jährlich mindestens 400.000 bis 600.000 nosokomiale Infektionen mit mindestens 10.000 bis 15.000 Todesfällen (Letalität 2,6 %; auf Intensivstationen bis zu 10 %) (1). Eine kontinuierliche Zunahme ist hier vor allem bei den multiresistenten gramnegativen Erregern (MRGN), insbesondere bei Bakterien aus der Familie der Enterobacteriaceae, zu verzeichnen. Diese werden in Deutschland seit 2012 nach ihrem Resistenzphänotyp als 3MRGN oder 4MRGN klassifiziert (2).
Aktuelle Epidemiologie
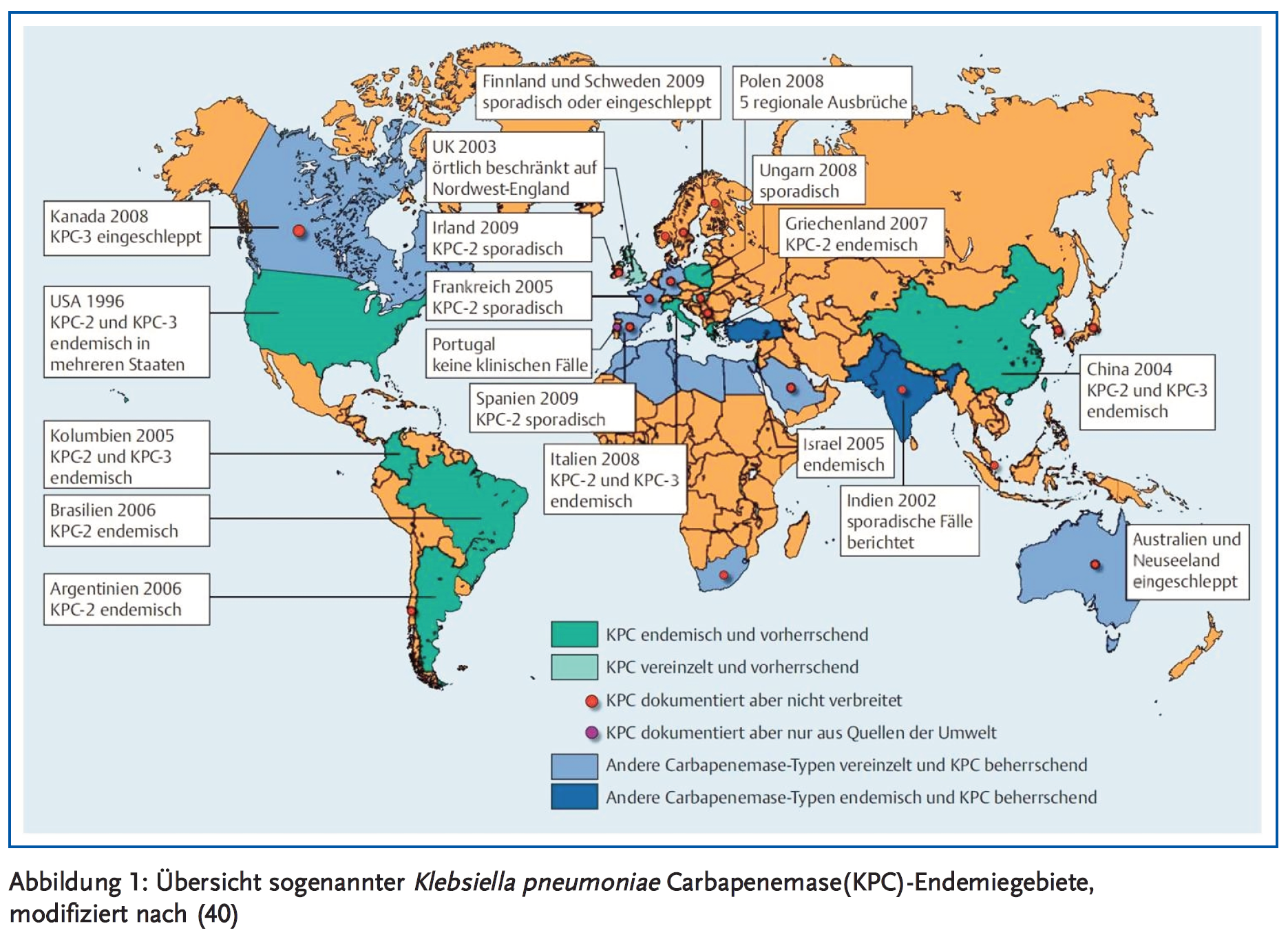
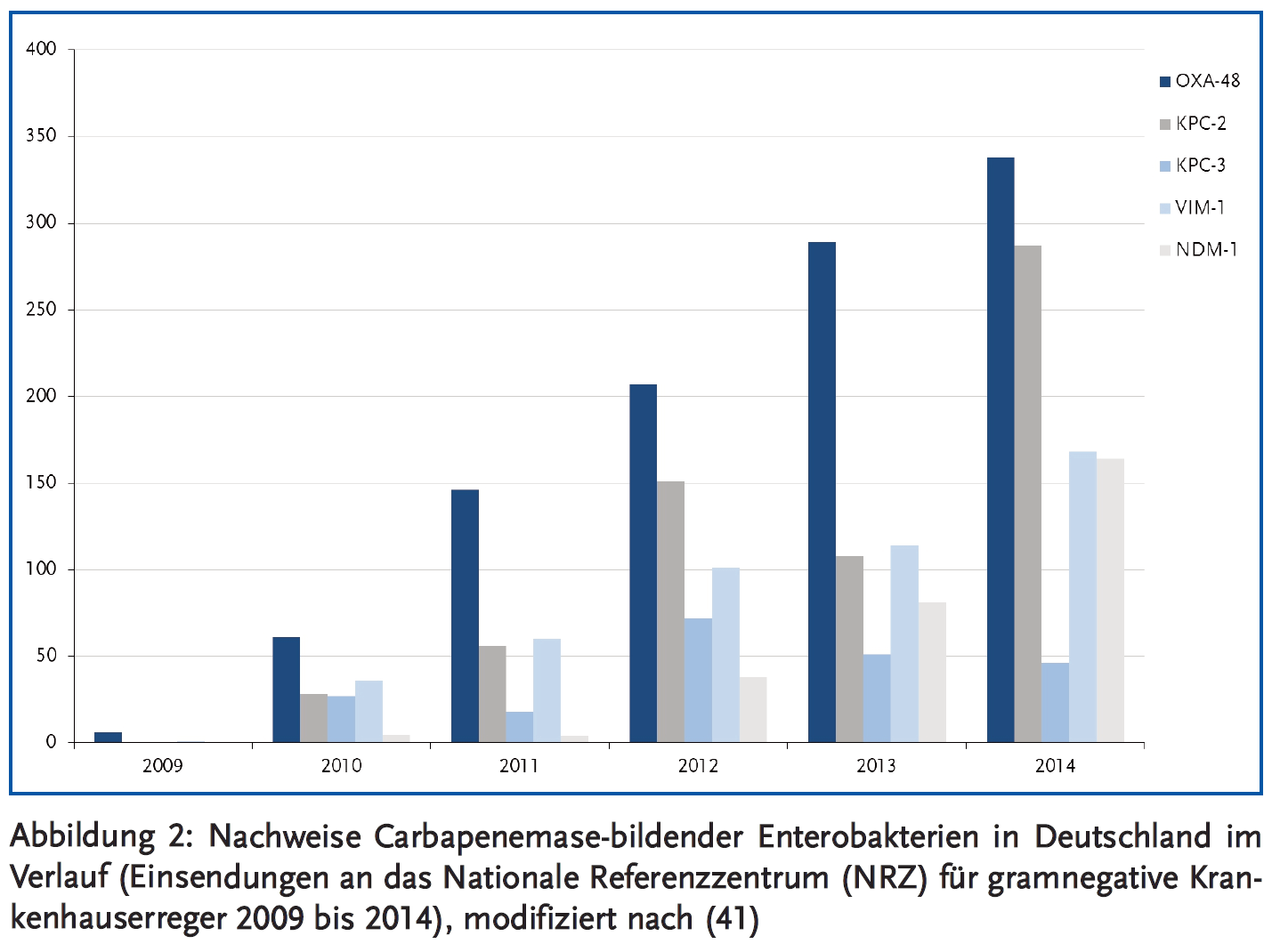
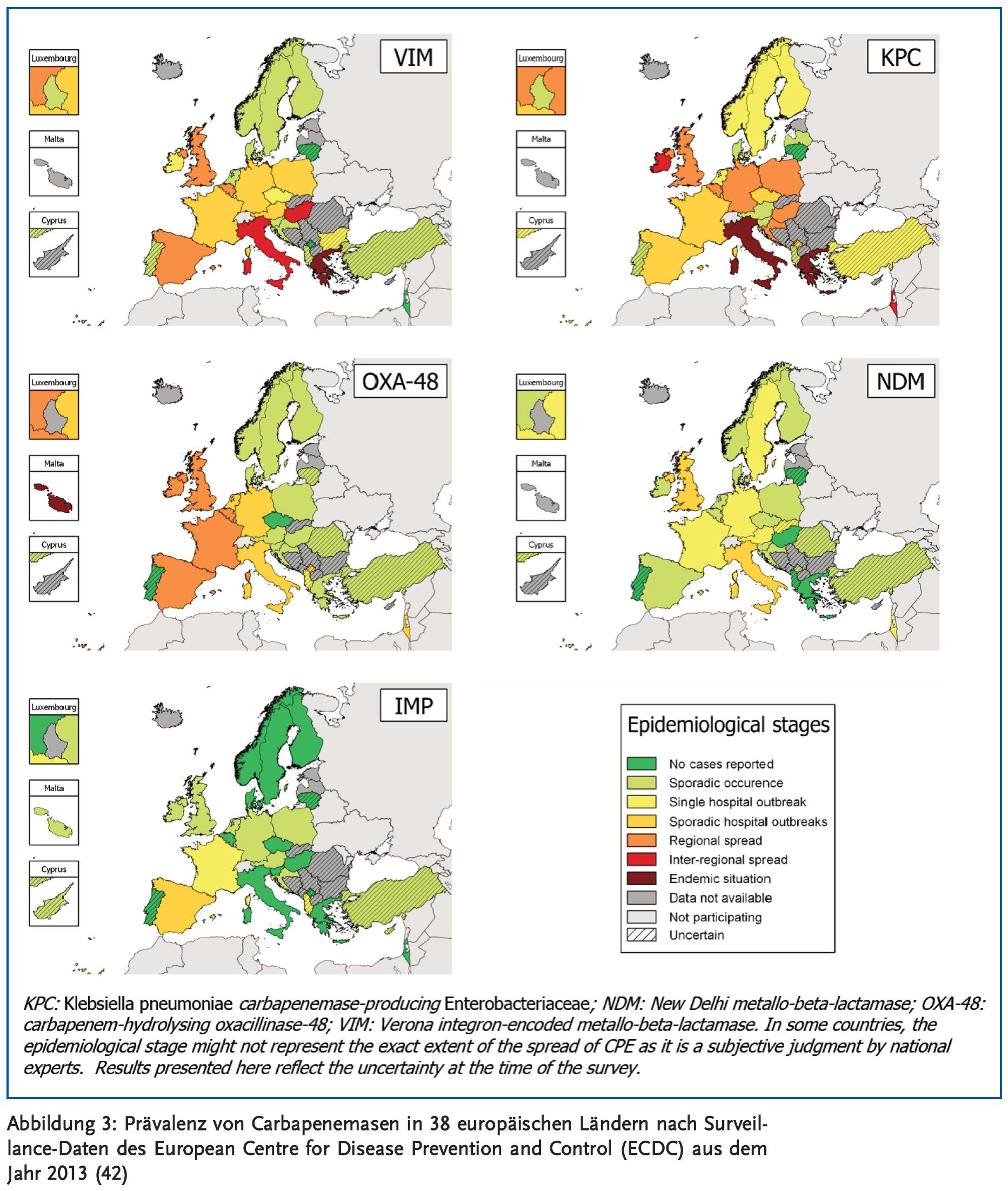
Enterobacteriaceae verursachen etwa 30 % aller Krankenhausinfektionen in Europa und in den USA (3). Mit dem Aufkommen von Extended-Spectrum-Beta-Lactamase(ESBL)-produzierenden Enterobakterien hat die Anwendung von Carbapenemen in den letzten Jahren dramatisch zugenommen (4). Daraus resultierend hat sich weltweit eine Zunahme von Carbapenem-Resistenzen ergeben, insbesondere bei Klebsiella pneumoniae. Obwohl diese Entwicklung lange Zeit weitgehend auf Krankenhäuser an der Ost- bzw. Nordostküste der USA beschränkt blieb (New York seit 2001), wurden Carbapenem-resistente K. pneumoniae mittlerweile in allen US-Bundesstaaten mit Ausnahme von Maine, Idaho und Alaska nachgewiesen (3). Carbapenem-resistente Enterobakterien (CRE) haben sich darüber hinaus endemisch in Teilen von Südamerika, Südeuropa (insbesondere Griechenland und Italien), Afrika (besonders Ägypten), dem Nahen Osten (vor allem Israel) und in Asien (vor allem Indien und Ostchina) etabliert, wodurch in der Folge immer wieder größere Ausbrüche vorkamen mit einem entsprechend relevanten internationalen Bedrohungsszenario (5) (Abbildung 1). Beispielsweise ist der Anteil der Carbapenemase-bildenden Klebsiella-Isolate in US-Krankenhäusern von < 1 % im Jahr 2001 auf 12 % im Jahr 2010 angestiegen (3). In Griechenland stieg die gleiche Zahl von < 1 % im Jahr 2001 auf rund 70 % im Jahr 2012 an, und in Italien von < 2 % im Jahr 2008 auf rund 35 % im Jahr 2013 (6). Die aktuelle Entwicklung in Deutschland ist graphisch in Abbildung 2 dargestellt. Die Prävalenz von Carbapenem-resistenten Klebsiella-Isolaten liegt hier noch unter 1 % (7).
Wichtige Mechanismen der Carbapenem-Resistenz
Eine Carbapenem-Resistenz entsteht bei Enterobacteriaceae entweder durch Produktion Carbapenem-hydrolysierender Enzyme (Carbapenemasen) oder sehr viel seltener durch den Verlust äußerer Membranporine in Kombination mit einer Überproduktion von AmpC-Betalaktamasen oder ESBL-Produktion (3;8). Alle Betalaktamasen (und somit auch Carbapenemasen) werden häufig nach ihrer Aminosäurehomologie im Ambler-Klassifikationssystem eingeteilt (9) (Tabelle 1). Betalaktamasen der Ambler-Klassen A, C und D haben Serin-Aminosäuren an ihren aktiven Stellen, während Klasse-B-Betalaktamasen ein Zink-Ion benötigen und daher als Metallo-Betalaktamasen (MBLs) bezeichnet werden (3;8;10).
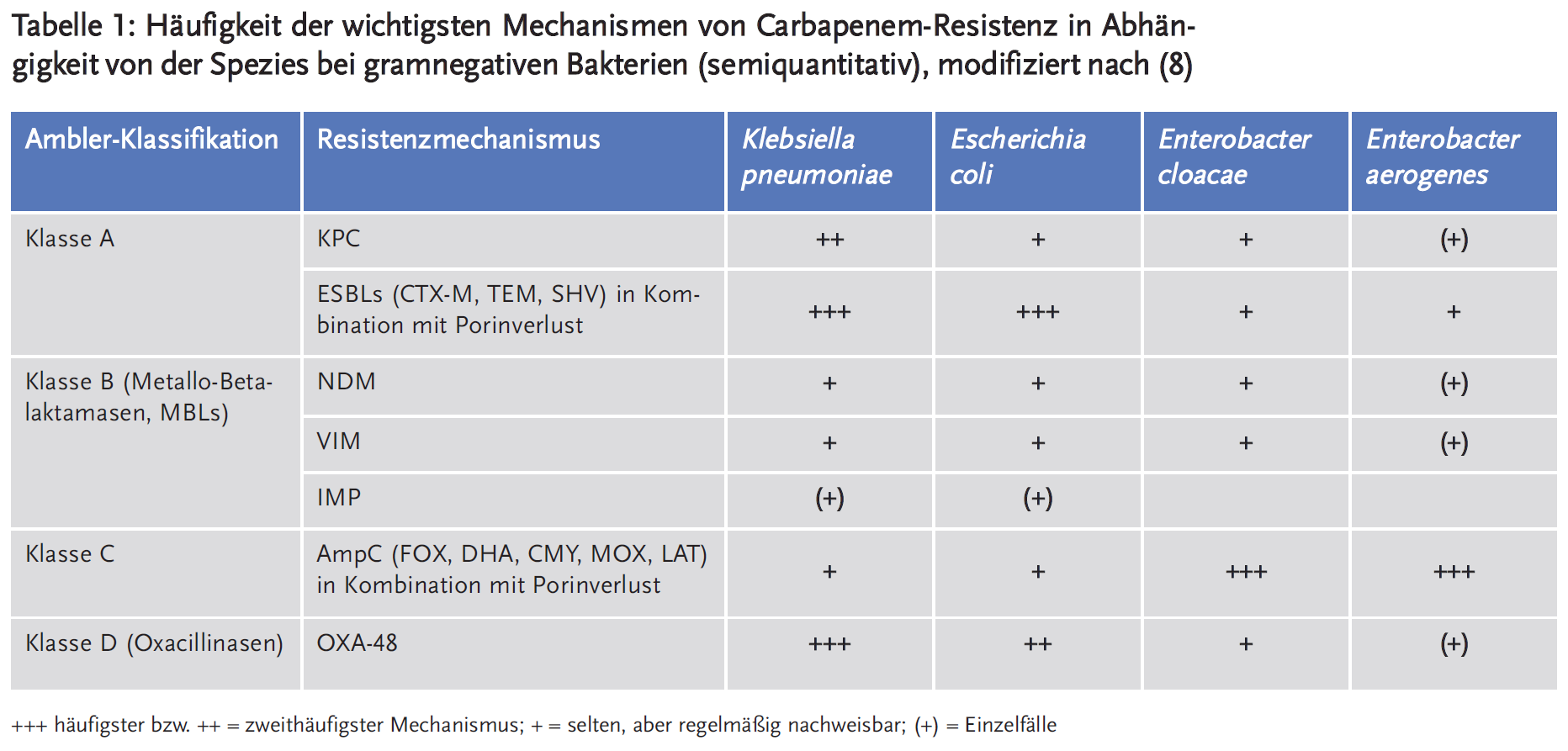
Klebsiella pneumoniae Carbapenemase (KPC) ist eine Klasse-A-Betalaktamase und stellt den derzeit dominierenden Resistenzmechanismus bei CRE in den USA, Südamerika, Südeuropa, Israel und China dar (3;8;10). Die verschiedenen KPC-Varianten hydrolysieren alle von der FDA und EMA zugelassenen Betalaktame und sind gegenüber den derzeit verfügbaren Betalaktamase-Inhibitoren einschließlich Clavulansäure, Sulbactam und Tazobactam stabil (8). KPC-produzierende Bakterien weisen darüber hinaus in der Regel weitere Resistenzen gegenüber anderen Antibiotikaklassen wie Fluorchinolonen und Aminoglykosiden auf, sodass nur wenige Behandlungsmöglichkeiten verbleiben (7;8;10). Das blaKPC-Gen wird plasmidisch kodiert und kann leicht innerhalb derselben Bakterienspezies, aber auch zwischen verschiedenen Spezies und Gattungen der Familie Enterobacteriaceae übertragen werden. Obwohl KPCs primär bei K. pneumoniae vorkommen, werden entsprechende Nachweise zunehmend auch bei Enterobacter spp., Citrobacter spp., Providencia spp., Morganella morganii, Serratia marcescens und Escherichia coli geführt (3;7;8;10).
MBLs hydrolysieren ebenfalls alle bekannten Betalaktamantibiotika bei Stabilität gegenüber derzeit verfügbaren Betalaktamase-Inhibitoren (3;8). Im Gegensatz zu KPCs können MBLs jedoch keine Monobaktame wie z. B. Aztreonam hydrolytisch aufspalten (8), sodass deren Wirksamkeit erhalten bleibt. Bis vor einigen Jahren waren Verona-Integron-Encoded-MBL (VIM) und selten Imipenemase (IMP) die bei Enterobakterien dominierenden MBL-Typen. Im Jahr 2009 jedoch wurde die ebenfalls plasmidisch kodierte New-Delhi-MBL (NDM) erstmals in Indien nachgewiesen und avancierte dank ihrer raschen Verbreitung innerhalb weniger Jahre zur häufigsten MBL in Indien, Pakistan und Großbritannien (5;8;11). Wie KPCs wird die NDM primär bei Klebsiella spp., aber auch bei verschiedenen anderen Spezies und Gattungen der Familie Enterobacteriaceae nachgewiesen (8;11).
Oxacillinasen (OXA) sind Klasse-D-Betalaktamasen, die nach ihrer Fähigkeit zur hydrolytischen Spaltung von Oxacillin benannt wurden. Innerhalb dieser Familie zeigen Enzyme vom OXA-48-Typ ausgeprägte Carbapenemase-Aktivität. Der Verbreitungsschwerpunkt von OXA-48-bildenden Bakterien liegt bislang in Südosteuropa, den Ländern Nordafrikas und in Indien (3;7;8;10).
Mikrobiologischer Nachweis von Carbapenemasen
Carbapenemasen lassen sich durch den Einsatz verschiedener Verfahren detektieren. Der molekulargenetische Nachweis des Resistenzmechanismus mittels PCR bei Anzucht eines Carbapenem-resistenten Isolates spielt im klinischen Alltag derzeit die größte Rolle. Bei Fehlen einer Carbapenemase ist die Bestimmung des Resistenzmechanismus meist sehr viel aufwändiger und epidemiologisch weniger bedeutsam (8). Eine Empfindlichkeitstestung auf Basis der minimalen Hemmkonzentration (MHK) des Erregers ist therapierelevant. Der direkte Einsatz von PCR-Techniken aus Originalmaterial vermag die „turn-around-time“ (TAT) erheblich zu verkürzen (< 24 Stunden) im Vergleich zur herkömmlichen kulturellen Anzüchtung (10), jedoch lassen sich hierbei nur bereits bekannte – und somit „testbare“ – Carbapenemasen erfassen.
Derzeit verfügbare antibiotische Therapieoptionen
Die verfügbaren Daten zur Behandlung von Infektionen durch CRE stammen meist aus retrospektiven oder nicht randomisierten Studien. Dementsprechend ist die Evidenz für bestimmte Empfehlungen begrenzt. Bei klinisch schwer verlaufenden CRE-Infektionen wird in der klinischen Praxis überwiegend mit einer Kombination aus unterschiedlichen Antibiotika therapiert (8). Eine kürzlich veröffentlichte Metaanalyse von 20 Studien zu Kombinationstherapien (u. a. von Tigecyclin mit Aminoglykosiden, Tigecyclin mit Colistin sowie Carbapenemen mit Colistin) bei über 700 Patienten (überwiegend Infektionen durch Klebsiella spp.) weist auf die Überlegenheit einer Kombinationstherapie gegenüber einer Monotherapie hin (12). In einer zweiten Metaanalyse, die überwiegend Kombinationstherapien von Colistin mit Carbapenemen, Tigecyclin, Aminoglykosiden oder auch dem Betalaktamase-Inhibitor Sulbactam untersuchte, ließ sich eine Überlegenheit der untersuchten Kombinationen gegenüber einer Monotherapie nicht zeigen (13).
Die derzeit verfügbaren Antibiotika mit hinreichender Aktivität gegen CRE sind Polymyxine (Colistin und Polymyxin B), Tigecyclin, Fosfomycin, Gentamicin und Amikacin (Tabelle 2). Jede dieser Substanzen weist erhebliche Einschränkungen und relevante Nebenwirkungen auf (3;7;8). Polymyxine sind mit Nephrotoxizitätsraten von 43 % bis 60 % behaftet und weisen zusätzlich eine klinisch relevante Neurotoxizität auf (14;15). Ihre optimale Dosierung ist weitgehend unklar, da pharmakokinetische und pharmakodynamische Eigenschaften erst vor Kurzem besser charakterisiert wurden (16;17). Herkömmliche Techniken für die mikrobiologische Empfindlichkeitsprüfung wie Agardiffusionstest und Epsilometer-Test (E-Test) sind für die Testung von Polymyxinen nicht unproblematisch, da es sich hierbei um relativ große Moleküle handelt, die schlecht in den verwandten Agar diffundieren und auf Glas- bzw. Kunststoffoberflächen binden, wodurch die In-vitro-Konzentrationen niedriger ausfallen als tatsächlich erwartet (18). Darüber hinaus gibt es bei vielen KPC-produzierenden Erregern Subpopulationen, die resistent gegen Colistin sind (sogenannte Heteroresistenz) (19). Aus klinischer Sicht weisen Polymyxine daher oftmals Wirkdefizite auf, insbesondere bei Unterdosierung (20). Mehrere Beobachtungsstudien lassen den Schluss zu, dass die Behandlung von Infektionen mit einer Polymyxin-Monotherapie ungünstigere klinische Ergebnisse liefert als die mit Betalaktamantibiotika, auch nach Adjustierung für sogenannte „confounders“ (3;7;8;20).
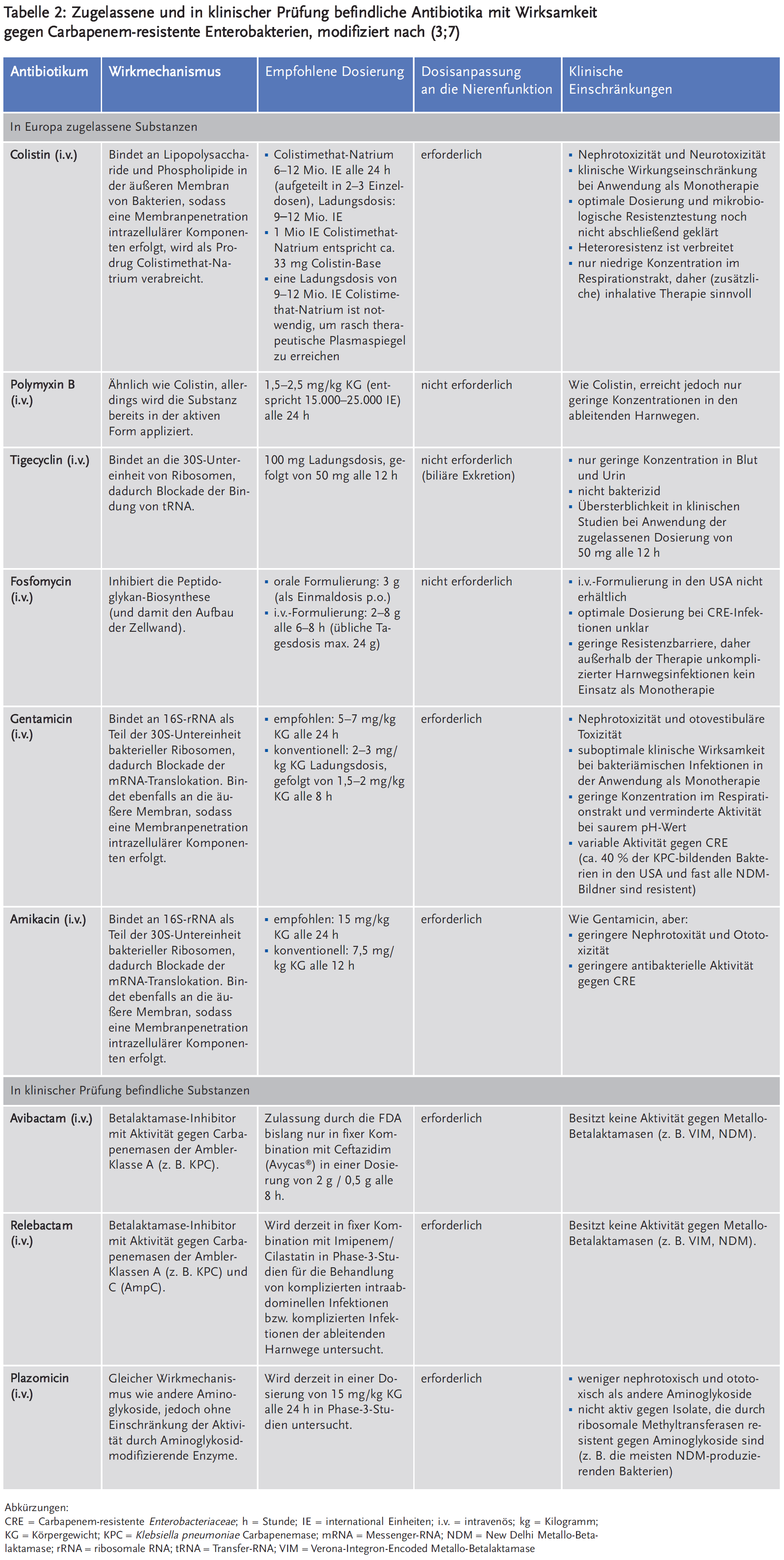
Für Tigecyclin gelten hinsichtlich der Behandlung von invasiven CRE-Infektionen noch gravierendere Vorbehalte. Tigecyclin ist nicht bakterizid (21), sodass seine Wirksamkeit gerade bei abwehrgeschwächten Patienten als eingeschränkt betrachtet werden muss, und es besitzt keine Aktivität gegenüber Pseudomonas aeruginosa (8). Die Anwendung von Tigecyclin geht mit niedrigen Plasmaspiegeln und geringen Harnwegskonzentrationen einher, woraus unzureichende Behandlungserfolge bei bakteriämischen Infektionen und Harnwegsinfektionen resultieren (21). Die Zulassung von Tigecyclin durch die FDA und die europäische Arzneimittelbehörde EMA umfasst die Therapie von komplizierten Infektionen der Haut-/Weichgewebe, intraabdominellen Infektionen sowie ambulant erworbenen Pneumonien. Selbst bei Anwendung für diese Indikationen zeigen sich innerhalb von randomisierten Studien eine erhöhte Mortalität und geringere Heilungsraten im Vergleich zu anderen Antibiotika, insbesondere bei der Behandlung bakteriämischer Infektionen (3;22). Unter Berücksichtigung von Pharmakodynamik/Pharmakokinetik-Aspekten und verfügbaren klinischen Daten wird daher eine Erhöhung der üblichen Tagesdosis auf z. B. die doppelte Dosis diskutiert (8). Bislang ist eine Ladungsdosis von 100 mg i.v. am Tag 0 und eine Erhaltungsdosis von zweimal 50 mg i.v. an den nachfolgenden Tagen üblich. In einer dreiarmigen Studie ist mittlerweile die Hochdosistherapie in zwei unterschiedlichen Dosierungen (150 mg Ladungsdosis, gefolgt von 2 x 75 mg tgl. i.v. sowie einer Ladungsdosis von 200 mg, gefolgt von 2 x 100 mg tgl. i.v.) gegenüber Imipenem/Cilastatin bei nosokomialer Pneumonie verglichen worden (23). Dabei wurde die höchste klinische Heilungsrate von 85 % gegenüber 75 % bei Imipenem/Cilastatin mit einer täglichen Dosierung von 2 x 100 mg erreicht (8). Der Erreger konnte jedoch nur in wenigen Fällen gesichert werden, sodass aus dieser Phase-2-Studie keine konkreten Schlüsse abgeleitet werden können, die eine allgemeine Hochdosistherapie rechtfertigen (8).
Fosfomycin liegt in einer oralen und einer intravenösen Darreichungsform vor. Das orale Fosfomycin-Trometamol spielt nur in der Therapie von unkomplizierten Harnwegsinfektionen eine Rolle und wird für diese Indikation als Einmalgabe (3 g p.o.) empfohlen (8;24). Eine orale Therapie bei schwer verlaufenden Infektionen durch als empfindlich getestete Erreger ist nicht sinnvoll. Die intravenöse Formulierung von Fosfomycin wird wegen der guten Gewebegängigkeit häufig als synergistischer Kombinationspartner eingesetzt. Eine Monotherapie mit Fosfomycin i.v. hingegen kann zu einer raschen Resistenzentwicklung führen (8). Bei einer Infektion durch CRE wählt man eine eher hohe Dosis von 3 x 5–8 g i.v. Auch bei Fosfomycin wird analog zur Applikation von Colistin der Einsatz einer Ladungsdosis von z. B. 12 g i.v. diskutiert. Wegen des hohen Natriumgehalts von 14,5 mmol Na+ in 1 g Fosfomycin kann die Substanz jedoch nicht bei jedem intensivmedizinischen Patienten zum Einsatz kommen. Studien über eine Kombination von Fosfomycin mit Colistin bei fremdkörperassoziierten Infektionen oder von Fosfomycin mit einem Aminoglykosid bei P. aeruginosa-Infektionen lieferten Hinweise für eine indifferente bis additive Wirkung gegenüber einer Monotherapie (8).
Die Aktivität von Aminoglykosiden gegen CRE ist variabel. Isolate aus Israel und Italien weisen höhere Gentamicin-Empfindlichkeitsraten (> 90 %) auf (3;25) als Isolate aus den USA und Griechenland (13 % bis 61 %) (3;10;26). CRE sind in aller Regel besser mit Gentamicin behandelbar als mit Amikacin und werden fast immer resistent gegen Tobramycin getestet (3;7). NDM-produzierenden CRE sind in der Regel resistent gegen alle verfügbaren Aminoglykoside (11). Auch im Falle einer guten In-vitro-Aktivität gelten Aminoglykoside als suboptimale Therapieoptionen wegen ihrer hohen Nephrotoxizitätsrate und der otovestibulären Toxizität mit irreversibler Innenohrschwerhörigkeit (27). Zudem weisen sie eine schlechte Penetration ins Lungengewebe (8) und eine vergleichsweise geringe Wirksamkeit bei der Anwendung als Monotherapie für gramnegative Bakteriämien auf (8).
Zusätzlich zu den bereits genannten klinischen Einschränkungen müssen leider auch für Polymyxine, Tigecyclin, Fosfomycin und Aminoglykoside ansteigende Resistenzraten bei der Behandlung von CRE-Infektionen berücksichtigt werden (3;8). Daher erscheint die Verwendung von Kombinationstherapien sinnvoll und notwendig (3;7;8). In-vitro-Synergien gegen CRE sind insbesondere für Polymyxine und Carbapeneme dokumentiert (28;29), trotz Vorliegen einer Resistenz gegen Carbapeneme. In einer klinischen Beobachtungsstudie aus Italien bei 125 Patienten mit Bakteriämien durch KPC-produzierenden K. pneumoniae war die 30-Tage-Mortalitätsrate bei Patienten, die eine Kombinationstherapie erhielten, deutlich niedriger als bei der Anwendung von Monotherapien (34 % vs. 54 %) (25). Andere Studien haben diese Ergebnisse bestätigt, wobei am häufigsten ein Benefit für Polymyxin-Carbapenem-Kombinationstherapien gezeigt werden konnte (8;28). Der Einsatz von Carbapenemen bei Infektionen durch CRE mag zunächst ungewöhnlich wirken, aber nicht in allen Fällen liegen tatsächlich gleichmäßig hohe MHK-Werte für die einzelnen Carbapeneme (Imipenem, Meropenem, Doripenem und Ertapenem) vor (8). Nutzen sollte man eine Dosissteigerung sowie eine verlängerte Infusionsdauer als Option der Wirkverstärkung. Dabei müssen die therapeutische Breite, die physikalisch-chemische Stabilität der Infusionslösung, die erreichbaren Plasma- und Gewebespiegel und die gemessene MHK des Erregers berücksichtigt werden (8). Nach Modellrechnungen kann man auch bei gesteigerter renaler Clearance (150 ml/min) z. B. mit einer Tagesdosis von 4 g Meropenem verteilt auf vier Einzeldosen mit verlängerter Infusionsdauer (zwei bis vier Stunden) eine Plasmakonzentration erreichen, die für Erreger mit einer MHK von 4–8 mg/l (niedriggradige Resistenz) ausreichend sein kann (der aktuelle EUCAST(European Committee on Antimicrobial Susceptibility Testing)-Grenzwert für Resistenz bei der Behandlung mit 3 x 1 g Meropenem als Kurzinfusion liegt bei 2 mg/l) (8). Höhere Dosen (mindestens 6 g verteilt auf drei bis vier Einzeldosen mit verlängerter Infusionsdauer) wären notwendig bei Erregern mit einer MHK von 16–32 mg/l. Erreger mit höheren MHK-Werten müssen auch bei Dosissteigerung als nicht mehr effektiv mit Meropenem behandelbar gelten (8). Ein infektiologisches Konsil bzw. Rücksprache mit einem Experten für antimikrobielle Chemotherapie ist jedoch nötig, da viele Details – vor allem auch die therapeutische Breite und das Erreichen ausreichender Wirkspiegel im Zielkompartiment – zu beachten sind (7;8).
Die optimale Behandlung von Infektionen durch CRE, die resistent gegen alle genannten Antibiotika sind, ist gegenwärtig unklar. Ein in Griechenland verfolgter Ansatz bei Infektionen durch panresistente KPC-produzierende K. pneumoniae ist, Ertapenem entweder mit Imipenem/Cilastatin, Meropenem oder Doripenem zu kombinieren (3;7;8). Die Rationale für diese Kombination ist, dass KPC größere Affinität für Ertapenem als für andere Carbapeneme aufweist, sodass Ertapenem als „Enzymfänger“ primär der Bindung von KPC dienen soll, um die Verfügbarkeit für die Hydrolyse des anderen angewandten Carbapenems zu reduzieren. Dieser Ansatz hat sich in-vitro sowie im Mausmodell als wirksam erwiesen (30) und wurde bei der Behandlung von vier Patienten mit panresistenten KPC-produzierenden K. pneumoniae klinisch erfolgreich angewandt (31;32).
Neue Antibiotika mit Wirksamkeit gegen CRE
Die Pipeline der Neuentwicklung von Substanzen mit Wirkung gegen CRE ist überschaubar (3;7;8) (Tabelle 2). Nennenswert sind das neue Aminoglykosid Plazomicin (ACHN-490) sowie die neuen Betalaktamase-Inhibitoren Avibactam (NXL-104) und Relebactam (MK-7655), die in Kombination mit unterschiedlichen Betalaktamantibiotika getestet wurden und für die bereits Zulassungsverfahren angebahnt bzw. eingeleitet wurden (7;8). Für die fixe Kombination von Ceftazidim und Avibactam ließ sich in Phase-3-Studien eine ausgezeichnete In-vitro-Aktivität gegen KPC-bildende Enterobacteriaceae zeigen, sodass von der US Food and Drug Administration (FDA) kürzlich eine Zulassung erteilt wurde (33). Bemerkenswert ist, dass Avibactam leider keine Aktivität gegen MBLs wie VIM oder NDM besitzt (34;35). Das gleiche gilt für Relebactam, das in Phase 3 der klinischen Prüfung in einer festen Kombination mit Imipenem/Cilastatin getestet wurde (36). Plazomicin ist ein neues Aminoglykosid, das sich ebenfalls in Phase 3 der klinischen Prüfung befindet und eine Aktivität gegen CRE besitzt, wenn diese bereits resistent gegen herkömmliche Aminoglykoside aufgrund Aminoglykosid-modifizierender Enzyme sind (37). Allerdings ist Plazomicin nicht ausreichend gegen NDM-bildende Bakterien wirksam, die in der Regel ribosomale Methyltransferasen besitzen, die zur Resistenz gegen Aminoglykoside führen (3;7).
Fazit für die Praxis
Die bestehenden Grenzen der genannten, kurz vor der Zulassung stehenden Antibiotika sowie der Mangel an erfolgversprechenden experimentellen Substanzen verdeutlichen sehr einprägsam, wie wichtig eine Erhöhung von Investitionen in die klinische Antibiotikaforschung und die damit verbundene gesamtgesellschaftliche Verantwortung ist (38;39). Die Verbesserung der Heilungschancen von invasiven Infektionen durch CRE wird maßgeblich an die Entwicklung und Zulassung neuer Antibiotikaklassen geknüpft sein. Da die derzeit verfügbaren antibiotischen Therapiemöglichkeiten äußerst begrenzt sind, kommt stringent praktizierten Maßnahmen zur Infektionsprävention sowie sorgsam praktiziertem Antibiotic Stewardship (ABS) herausragende Bedeutung zu.
Interessenkonflikte
Der Autor erhielt Vortragshonorare bzw. Unterstützung für Kongressbesuche von den Firmen Astellas, InfectoPharm, MSD, Novartis, Pfizer sowie von der Falk Foundation.
Dieser Beitrag wurde ohne finanzielle Unterstützung Dritter verfasst.
Literatur
- Behnke M, Hansen S, Leistner R et al.: Nosocomial infection and antibiotic use: a second national prevalence study in Germany. Dtsch Arztebl Int 2013; 110: 627-633.
- Hygienemaßnahmen bei Infektionen oder Besiedlung mit multiresistenten gramnegativen Stäbchen. Empfehlungen der Kommission für Krankenhaushygiene und Infektionsprävention (KRINKO) beim Robert Koch-Institut (RKI). Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 2012; 55: 1311-1354.
- Satlin MJ, Jenkins SG, Walsh TJ: The global challenge of carbapenem-resistant Enterobacteriaceae in transplant recipients and patients with hematologic malignancies. Clin Infect Dis 2014; 58: 1274-1283.
- Pitout JD, Laupland KB: Extended-spectrum beta-lactamase-producing Enterobacteriaceae: an emerging public-health concern. Lancet Infect Dis 2008; 8: 159-166.
- Munoz-Price LS, Poirel L, Bonomo RA et al.: Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis 2013; 13: 785-796.
- Mouloudi E, Protonotariou E, Zagorianou A et al.: Bloodstream infections caused by metallo-beta-lactamase/Klebsiella pneumoniae carbapenemase-producing K. pneumoniae among intensive care unit patients in Greece: risk factors for infection and impact of type of resistance on outcomes. Infect Control Hosp Epidemiol 2010; 31: 1250-1256.
- Lubbert C, Hau HM, Rodloff A et al.: [Clinical impact of infections with carbapenem-resistant enterobacteriaceae in liver transplant recipients]. Z Gastroenterol 2015; 53: 1276-1287.
- Mischnik A, Kaase M, Lubbert C et al.: [Carbapenem-resistance in Enterobacteriaceae, Pseudomonas aeruginosa and Acinetobacter baumannii]. Dtsch Med Wochenschr 2015; 140: 172-176.
- Ambler RP: The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci 1980; 289: 321-331.
- Temkin E, Adler A, Lerner A, Carmeli Y: Carbapenem-resistant Enterobacteriaceae: biology, epidemiology, and management. Ann N Y Acad Sci 2014; 1323: 22-42.
- Kumarasamy KK, Toleman MA, Walsh TR et al.: Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis 2010; 10: 597-602.
- Falagas ME, Lourida P, Poulikakos P et al.: Antibiotic treatment of infections due to carbapenem-resistant Enterobacteriaceae: systematic evaluation of the available evidence. Antimicrob Agents Chemother 2014; 58: 654-663.
- Sbrana F, Malacarne P, Viaggi B et al.: Carbapenem-sparing antibiotic regimens for infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae in intensive care unit. Clin Infect Dis 2013; 56: 697-700.
- Pogue JM, Lee J, Marchaim D et al.: Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin Infect Dis 2011; 53: 879-884.
- Kubin CJ, Ellman TM, Phadke V et al.: Incidence and predictors of acute kidney injury associated with intravenous polymyxin B therapy. J Infect 2012; 65: 80-87.
- Garonzik SM, Li J, Thamlikitkul V et al.: Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 2011; 55: 3284-3294.
- Sandri AM, Landersdorfer CB, Jacob J et al.: Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin Infect Dis 2013; 57: 524-531.
- Hindler JA, Humphries RM: Colistin MIC variability by method for contemporary clinical isolates of multidrug-resistant Gram-negative bacilli. J Clin Microbiol 2013; 51: 1678-1684.
- Meletis G, Tzampaz E, Sianou E et al.: Colistin heteroresistance in carbapenemase-producing Klebsiella pneumoniae. J Antimicrob Chemother 2011; 66: 946-947.
- Paul M, Bishara J, Levcovich A et al.: Effectiveness and safety of colistin: prospective comparative cohort study. J Antimicrob Chemother 2010; 65: 1019-1027.
- MacGowan AP: Tigecycline pharmacokinetic/pharmacodynamic update. J Antimicrob Chemother 2008; 62 Suppl 1: i11-i16.
- Prasad P, Sun J, Danner RL, Natanson C: Excess deaths associated with tigecycline after approval based on noninferiority trials. Clin Infect Dis 2012; 54: 1699-1709.
- Ramirez J, Dartois N, Gandjini H et al.: Randomized phase 2 trial to evaluate the clinical efficacy of two high-dosage tigecycline regimens versus imipenem-cilastatin for treatment of hospital-acquired pneumonia. Antimicrob Agents Chemother 2013; 57: 1756-1762.
- Karageorgopoulos DE, Wang R, Yu XH, Falagas ME: Fosfomycin: evaluation of the published evidence on the emergence of antimicrobial resistance in Gram-negative pathogens. J Antimicrob Chemother 2012; 67: 255-268.
- Tumbarello M, Viale P, Viscoli C et al.: Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: importance of combination therapy. Clin Infect Dis 2012; 55: 943-950.
- Daikos GL, Markogiannakis A: Carbapenemase-producing Klebsiella pneumoniae: (when) might we still consider treating with carbapenems? Clin Microbiol Infect 2011; 17: 1135-1141.
- Smith CR, Lipsky JJ, Laskin OL et al.: Double-blind comparison of the nephrotoxicity and auditory toxicity of gentamicin and tobramycin. N Engl J Med 1980; 302: 1106-1109.
- Qureshi ZA, Paterson DL, Potoski BA et al.: Treatment outcome of bacteremia due to KPC-producing Klebsiella pneumoniae: superiority of combination antimicrobial regimens. Antimicrob Agents Chemother 2012; 56: 2108-2113.
- Jernigan MG, Press EG, Nguyen MH et al.: The combination of doripenem and colistin is bactericidal and synergistic against colistin-resistant, carbapenemase-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 2012; 56: 3395-3398.
- Bulik CC, Nicolau DP: Double-carbapenem therapy for carbapenemase-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 2011; 55: 3002-3004.
- Giamarellou H, Galani L, Baziaka F, Karaiskos I: Effectiveness of a double-carbapenem regimen for infections in humans due to carbapenemase-producing pandrug-resistant Klebsiella pneumoniae. Antimicrob Agents Chemother 2013; 57: 2388-2390.
- Ceccarelli G, Falcone M, Giordano A et al.: Successful ertapenem-doripenem combination treatment of bacteremic ventilator-associated pneumonia due to colistin-resistant KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 2013; 57: 2900-2901.
- Zhanel GG, Lawson CD, Adam H et al.: Ceftazidime-avibactam: a novel cephalosporin/beta-lactamase inhibitor combination. Drugs 2013; 73: 159-177.
- Lucasti C, Popescu I, Ramesh MK et al.: Comparative study of the efficacy and safety of ceftazidime/avibactam plus metronidazole versus meropenem in the treatment of complicated intra-abdominal infections in hospitalized adults: results of a randomized, double-blind, Phase II trial. J Antimicrob Chemother 2013; 68: 1183-1192.
- Vazquez JA, Gonzalez Patzan LD, Stricklin D et al.: Efficacy and safety of ceftazidime-avibactam versus imipenem-cilastatin in the treatment of complicated urinary tract infections, including acute pyelonephritis, in hospitalized adults: results of a prospective, investigator-blinded, randomized study. Curr Med Res Opin 2012; 28: 1921-1931.
- Toussaint KA, Gallagher JC: beta-lactam/beta-lactamase inhibitor combinations: from then to now. Ann Pharmacother 2015; 49: 86-98.
- Livermore DM, Mushtaq S, Warner M et al.: Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J Antimicrob Chemother 2011; 66: 48-53.
- Boucher HW, Talbot GH, Benjamin DK, Jr. et al.: 10 x '20 Progress-development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis 2013; 56: 1685-1694.
- Akademie der Wissenschaften in Hamburg, Deutsche Akademie der Naturforscher Leopoldina (Hrsg.): Antibiotika-Forschung: Probleme und Perspektiven – Stellungnahme. Band 2, Abhandlungen der Akademie der Wissenschaften in Hamburg. Berlin, Boston: Walter de Gruyter, 2013.
- Lübbert C: Was lernen wir aus dem Leipziger KPC-Ausbruch? Krankenhaushygiene up2date 2014; 8 (1): 13-20.
- Bericht des Nationalen Referenzzentrums für gramnegative Krankenhauserreger (1. Januar 2013 bis 31. Dezember 2013). Epidemiol Bull 2014; Nr. 43: 421-425.
- European Centre for Disease Prevention and Control: Antimicrobial resistance surveillance in Europe 2013. Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net). Stockholm: ECDC; 2014.