Remdesivir (Veklury®) ▼ (2021)
Zugelassene Indikation und Wirkmechanismus
Veklury® (Remdesivir) ist bedingt zugelassen zur Behandlung von COVID-19 bei Erwachsenen und Jugendlichen (im Alter von mindestens 12 Jahren und mit einem Körpergewicht von mindestens 40 kg) mit einer Pneumonie, die eine zusätzliche Sauerstoffzufuhr erfordert (Low- oder High-Flow-Sauerstoff oder nicht-invasive Beatmung zu Beginn der Behandlung).
Remdesivir ist ein Adenosin-Nukleotid-Prodrug, das in Wirtszellen zum wirksamen Nukleosid-Triphosphat-Metaboliten umgewandelt wird. Remdesivir-Triphosphat wirkt als ein Analogon von Adenosin-Triphosphat (ATP) und konkurriert mit dem natürlichen ATP-Substrat um die Integration in entstehende RNA-Ketten durch die SARS-CoV-2-RNA-abhängige RNA-Polymerase. Dies führt zu einer verzögerten Kettenterminierung während der Replikation der viralen RNA und damit zur Hemmung der Virusreplikation.
Markteinführung
Veklury® (Remdesivir) ist seit 1. Juni 2021 für den stationären Gebrauch über krankenhausversorgende Apotheken sowie Krankenhausapotheken auf dem deutschen Markt verfügbar.
Bewertung
Remdesivir erhielt am 3. Juli 2020 eine sogenannte bedingte Zulassung („conditional marketing authorisation“, CMA) zur Behandlung sauerstoffpflichtiger Patienten mit COVID-19-Pneumonie. Die CMA wurde auf Grundlage vorläufiger Daten aus der Studie NIAID-ACTT-1 erteilt. Eine Neubewertung der CMA erfolgte im Dezember 2020. Hierbei wurden in erster Linie die finalen Ergebnisse zur 28-Tage-Mortalität in der NIAID-ACTT-1-Studie berücksichtigt. Ergänzend wurden die Zwischenergebnisse der WHO-Studie SOLIDARITY herangezogen.
In der Studie NIAID-ACTT-1 war in der Gesamtgruppe die Zeit bis zur Genesung (operationalisiert als Zeit bis zur Entlassung aus stationärer Behandlung) unter Remdesivir signifikant verkürzt (10 vs. 15 Tage). Bezüglich der 28-Tage-Mortalität bestand in der Gesamtgruppe lediglich ein numerischer Vorteil unter Remdesivir (11,4 % vs. 15,2 %, nicht signifikant). Subgruppenanalysen zeigten eine deutlich unterschiedliche Effektivität von Remdesivir in Abhängigkeit von der Krankheitsschwere bei Behandlungsbeginn: Patienten mit Low-Flow-Sauerstofftherapie hatten hinsichtlich beider Endpunkte einen signifikanten Vorteil unter Remdesivir, dagegen war bei Patienten mit invasiver Beatmung oder ECMO (extrakorporaler Membranoxygenierung) sowohl die Mortalität numerisch erhöht als auch die Zeit bis zur Genesung numerisch verlängert. Diese Ergebnisse werden bestätigt durch Subgruppenanalysen der WHO-Studie SOLIDARITY, die einen negativen Trend auf die krankenhausinterne Mortalität bei Patienten zeigen, die zu Studienbeginn beatmet wurden. Pathophysiologisch erscheint es plausibel, dass der weitere Krankheitsverlauf durch Remdesivir nicht mehr positiv beeinflusst werden kann, wenn bei Behandlungsbeginn bereits die systemische Entzündungsreaktion im Vordergrund steht.
Insgesamt erscheint Remdesivir gut verträglich. Subgruppenanalysen deuten auf nephrotoxische und hepatotoxische Effekte bei Risikopatienten hin. Die Langzeitsicherheit von Remdesivir kann noch nicht beurteilt werden.
In der Zwischenzeit wurde die Zulassung von Remdesivir auf Patienten mit COVID-19-Pneumonie eingeschränkt, die bei Behandlungsbeginn Low- oder High-Flow-Sauerstoff oder eine nicht-invasive Beatmung (NIV) benötigen. Aus Sicht der AkdÄ ist jedoch das Nutzen-Risiko-Verhältnis von Remdesivir bei Patienten mit High-Flow-Sauerstofftherapie oder NIV kritisch zu sehen (siehe auch „Neue Arzneimittel“ Remdesivir aus dem Jahr 2020 (1)). Diese Patientengruppe ist in der Studie NIAID-ACTT zu klein, um eine valide Aussage zur Effektivität abzuleiten; numerisch besteht allenfalls ein Nutzen bezüglich der Zeit bis zur Genesung, nicht bezüglich der Mortalität. Diesem unsicheren Nutzen stehen Warnsignale für nephrotoxische und hepatotoxische Effekte gegenüber. Die aktuelle S3-Leitlinie zur stationären Behandlung von COVID-19-Patienten verzichtet derzeit auf eine Empfehlung für oder gegen den Einsatz von Remdesivir (2). Nach Einschätzung der AkdÄ ist aktuell der Einsatz von Remdesivir allenfalls bei COVID-19-Patienten mit Low-Flow-Sauerstoffbedarf gerechtfertigt, nicht aber bei Patienten mit High-Flow-Sauerstofftherapie, NIV oder invasiver Beatmung einschließlich ECMO. Der therapeutische Stellenwert ist unklar, weitere klinische Studien sind erforderlich.
Wirksamkeit in den Zulassungsstudien
Die Studie NIAID-ACTT-1 (3) ist eine doppelblinde, randomisierte Studie zum Vergleich von Remdesivir und Placebo bei hospitalisierten Patienten mit COVID-19 Infektion (n = 1062). Eingeschlossen wurden sowohl Patienten mit akuter respiratorischer Insuffizienz (Sauerstoffsättigung (SpO2) ≤ 94 % oder Notwendigkeit einer Sauerstoffgabe oder maschineller Beatmung) als auch respiratorisch stabile Patienten mit radiologischem Verdacht auf Lungeninfiltrate. Patienten mit Niereninsuffizienz (eGFR < 30 ml/min) oder erhöhten Transaminasen (Alanin-Aminotransferase [ALT] > 5 ULN) wurden ausgeschlossen.
Es erfolgte eine Randomisierung 1:1 zu Remdesivir (200 mg an Tag 1, gefolgt von täglich 100 mg) oder Placebo zusätzlich zur zentrumsüblichen Therapie („standard of care“). Lediglich 38 % (Remdesivir-Arm) bzw. 43 % (Placebo-Arm) der Patienten erhielten die Studienmedikation wie vorgesehen über zehn Tage. Die häufigste Begleitmedikation bestand in Hydroxychloroquin (36 %), ein Viertel der Patienten erhielt Dexamethason (siehe unten).
15 % der Patienten stammten aus Europa, der überwiegende Anteil (80 %) aus Nordamerika. Das mittlere Alter lag bei 59 Jahren. Etwa zwei Drittel der Patienten waren männlich. Der Symptombeginn lag im Median neun Tage vor Randomisierung. Bei der Mehrzahl der Patienten lagen Vorerkrankungen vor (50 % Hypertonus, 37 % Adipositas, 30 % Diabetes mellitus Typ 2, 11 % Asthma bronchiale).
Primärer Endpunkt der Studie NIAID-ACTT-1 war die Zeit bis zur Genesung, operationalisiert als Zeit bis zur Entlassung aus stationärer Behandlung, unabhängig vom Zustand des Patienten. Die Analyse erfolgte stratifiziert nach der Krankheitsschwere zum Zeitpunkt der Randomisierung (mild/moderat vs. schwer). Patienten ohne Sauerstoffbedarf wurden entsprechend ihrer Atemfrequenz entweder der mild/moderat erkrankten Gruppe (bei Atemfrequenz < 24/min) oder der schwer erkrankten Gruppe (Atemfrequenz ≥ 24/min) zugeordnet. 90 % der Patienten waren zum Zeitpunkt der Randomisierung „schwer“ erkrankt, definiert als notwendige maschinelle Beatmung, notwendige Sauerstoffgabe, SpO2 ≤ 94 % oder Tachypnoe mit einer Atemfrequenz ≥ 24/min (siehe Tabelle 1).
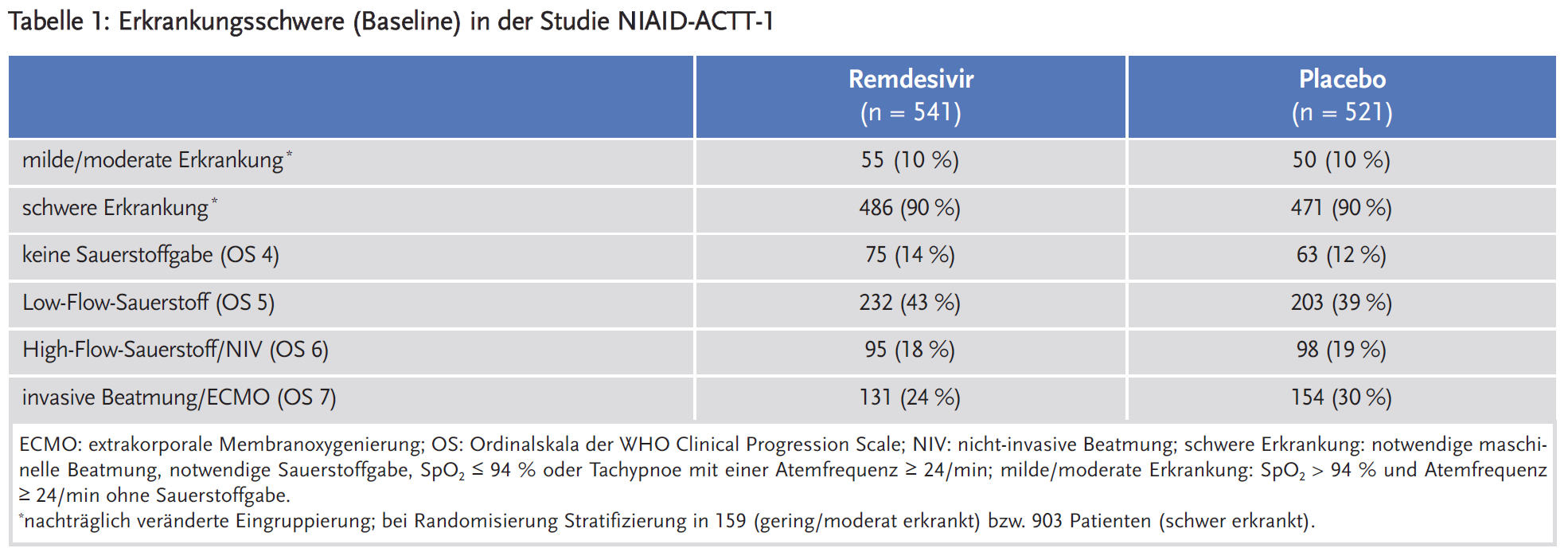
Wie bereits 2020 in „Neue Arzneimittel“ zu Remdesivir (1) kritisiert, entspricht die dichotome Aufteilung der Krankheitsschwere nicht der gängigen klinischen Praxis. Dagegen korreliert die Ordinalskala der WHO („Clinical Progression Scale“ (4)) sowohl mit typischen klinischen Behandlungssettings (Score 4: Normalstation, Score 5: Intermediate Care, Score ≥ 6: Intensivstation) als auch mit pathophysiologisch unterschiedlichen Erkrankungsstadien.
In der Gesamtgruppe und bei Patienten mit „schwerer Erkrankung“ war die mediane Zeit bis zur Genesung unter Remdesivir signifikant verkürzt (10 vs. 15 Tage bzw. 11 vs. 18 Tage). Bei Patienten mit „leichter oder moderater Erkrankung“ zeigte sich kein Behandlungsunterschied. Bei einer Subgruppenanalyse entsprechend der Clinical Progression Scale war der Behandlungsunterschied nur für Patienten mit Low-Flow-Sauerstofftherapie (Score 4) statistisch signifikant. Unter Remdesivir war hier allerdings die Zeit bis zur Genesung lediglich im Median um zwei Tage verkürzt (7 vs. 9 Tage).
Für den wichtigen sekundären Endpunkt der 28-Tage-Mortalität ist die Subgruppenanalyse nicht präspezifiziert. Eine explorative Analyse kommt jedoch zu konsistenten Ergebnissen: Während sich in der Gesamtgruppe ein Trend zu einer reduzierten Mortalität unter Remdesivir zeigt (11,4 % vs. 15,2 %; Hazard Ratio [HR] 0,73; 95 % Konfidenzintervall [CI] 0,52–1,03), besteht in der Subgruppenanalyse nur für Patienten mit Low-Flow-Sauerstofftherapie ein Vorteil unter Remdesivir. In der Gruppe der Patienten mit invasiver Beatmung oder ECMO findet sich sogar ein Trend zu einer erhöhten Mortalität unter Remdesivir (siehe Tabelle 2). In Übereinstimmung mit diesen Ergebnissen zeigen Interaktionstests bei Patienten mit Score 6 und 7 der Clinical Progression Scale eine reduzierte Wirksamkeit von Remdesivir auf die Zeit bis zur Genesung und auf die Mortalität.
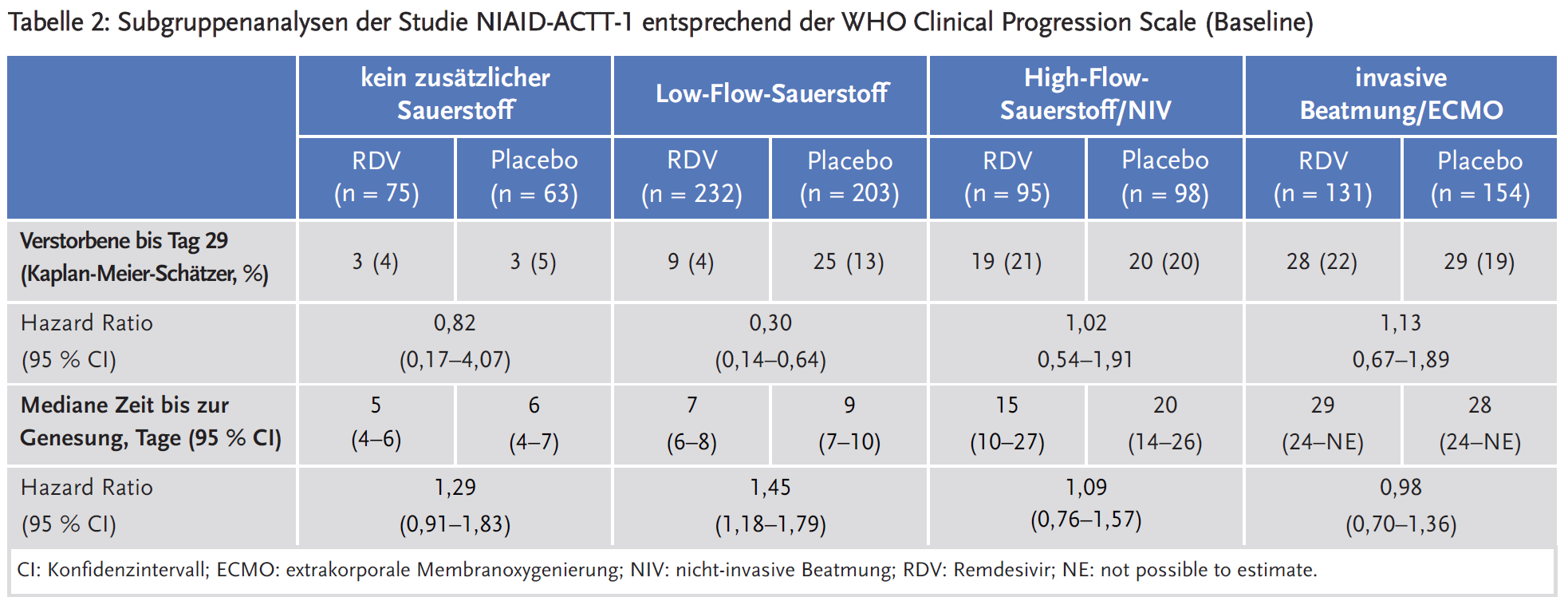
Die Ergebnisse der Studie NIAID-ACTT-1 werden bestätigt durch die Zwischenresultate der WHO-Studie SOLIDARITY (5), einer randomisierten, unverblindeten, adaptiven Studie. Die Studie verglich bei stationär behandelten Patienten mit gesicherter Covid-19-Erkrankung mehrere Arzneimittel, darunter Remdesivir (n = 2750), mit dem Standard of Care (n = 2725 als Kontrolle zu Remdesivir). Mit nur 9 % waren deutlich weniger Patienten als in der Studie NIAID-ACTT-1 beatmet. Dabei wurde nicht erfasst, ob die Beatmung invasiv oder nicht-invasiv erfolgte. In der Gesamtgruppe bestand kein eindeutiger Effekt von Remdesivir auf die krankenhausinterne Mortalität (Relatives Risiko [RR] 0,95; 95 % CI 0,81–1,11). Subgruppenanalysen zeigten einen negativen Trend auf die Mortalität bei Patienten, die zu Studienbeginn beatmet wurden (RR 1,20; 95 % CI 0,80–1,80), im Vergleich zu nicht-beatmeten Patienten (RR 0,86; 95 % CI 0,67–1,11).
Eine Metaanalyse der EMA schloss neben den Studien NIAID-ACTT-1 und SOLIDARITY auch zwei kleinere Studien ein: Eine randomisierte, doppelblinde Studie aus China (n = 237), die vor Erreichen der ursprünglich angestrebten Fallzahl abgebrochen wurde (6), sowie eine randomisierte, unverblindete Studie mit Einschluss von moderat erkrankten Patienten, die entweder keinen zusätzlichen Sauerstoffbedarf hatten oder lediglich eine Low-Flow-Sauerstofftherapie erhielten (n = 596) (7). Die Aussagekraft der Metaanalyse ist eingeschränkt durch die starke Heterogenität der eingeschlossenen Studien. Die Ergebnisse bestätigen jedoch den unterschiedlichen Behandlungseffekt von Remdesivir in Abhängigkeit von dem Beatmungsstatus der Patienten: Die gepoolte Analyse zeigte in der Gesamtpopulation eine nichtsignifikante Reduktion der Mortalität unter Remdesivir; in der Subgruppenanalyse bestand dieser positive Trend lediglich bei Patienten, die bei Studienbeginn nicht beatmet wurden.
Die Begleitmedikation mit Dexamethason war in den Studien NIAID-ACTT-1 und SOLIDARITY zwischen den Studienarmen ausgeglichen, wobei deutlich weniger Patienten in der Studie NIAID-ACTT-1 als in der Studie SOLIDARITY Dexamethason erhielten (23 % vs. 48 %). Es liegen in beiden Studien keine Angaben zur Dosierung von Dexamethason vor. Vorliegende Sensitivitäts- und Subgruppenanalysen stratifizieren nicht zwischen der Verwendung von Kortikosteroiden bei Studienbeginn und ihrer Verwendung im weiteren Studienverlauf. Es ist anzunehmen, dass die Kortikosteroidbehandlung bevorzugt bei Patienten mit einem schlechteren klinischen Status eingeleitet wurde. So war in der Studie SOLIDARITY in beiden Gruppen der Einsatz von Kortikosteroiden mit einer höheren Mortalität assoziiert (Remdesivir: 16,6 % vs. 8,0 %; Placebo: 17,9 % vs. 6,9 %). Insgesamt lassen sich aus den vorliegenden Daten keine sicheren Aussagen zur Begleitmedikation mit Dexamethason ableiten.
Ausgewählte Nebenwirkungen
Unter Remdesivir traten bei gesunden Probanden gehäuft Transaminasenerhöhungen auf (14 %). Bei Patienten mit COVID-19 bestand diesbezüglich kein relevanter Unterschied zum Placebo-Arm (zur potenziellen Hepatotoxizität siehe unten). Häufige Nebenwirkungen waren außerdem Kopfschmerzen, Übelkeit und Hautreaktionen. Überempfindlichkeitsreaktionen unter Remdesivir waren selten.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
- Der antivirale Wirkmechanismus und die Erfahrungen mit Neuraminidasehemmern bei Influenza sprechen für eine geringere Effektivität von Remdesivir bei spätem Behandlungsbeginn. Dies wird gestützt durch eine Subgruppenanalyse der Studie NIAID-ACTT-1: Hier zeigte sich ein schwächerer Effekt von Remdesivir bei einem Behandlungsbeginn mehr als zehn Tage nach Symptombeginn.
- Aufgrund der in vitro beobachteten antagonistischen Wirkung wird die gleichzeitige Anwendung von Remdesivir und Chloroquinphosphat oder Hydroxychloroquinsulfat nicht empfohlen.
- Remdesivir wird überwiegend renal eliminiert. Es liegen keine Daten zur Effektivität und Sicherheit bei Patienten mit einer eGFR < 30 ml/min vor. Remdesivir sollte deshalb bei einer eGFR < 30 ml/min nicht angewendet werden.
- In tierexperimentellen Studien wurde eine schwere Nierentoxizität beobachtet. Der Mechanismus und die Relevanz für den Menschen sind unklar. Eine kumulative Exposition von Remdesivir könnte mit einem erhöhten Risiko für Nierenschäden assoziiert sein: Bei kritisch erkrankten Patienten (WHO Score 7) mit langer Therapiedauer wurde in der Studie NIAID-ACTT-1 unter Remdesivir häufiger die Behandlung aufgrund renaler Nebenwirkungen abgebrochen (13,6 % vs. 9,2 %).
- In der Studie NIAID-ACTT-1 waren in der Kontrollgruppe chronische Lebererkrankungen nicht mit einer erhöhten Mortalität assoziiert. Dagegen war im Remdesivir-Arm die Mortalität numerisch deutlich erhöht bei Patienten mit chronischen Lebererkrankungen (24,4 % vs. 12,3 %). Aufgrund der kleinen Fallzahlen lassen sich hieraus keine sicheren Aussagen ableiten. Remdesivir sollte jedoch bei Patienten mit chronischen Lebererkrankungen mit besonderer Vorsicht eingesetzt werden. Die Leberfunktion ist regelmäßig während der Therapie zu kontrollieren. Sollte unter Remdesivir ein fortgesetzter Anstieg beobachtet werden, ist die Anwendung zu pausieren oder zu beenden. Bei einem ALT-Wert > 5 ULN soll Remdesivir nicht angewendet werden, da für diese Patientengruppe keine Studiendaten vorliegen.

Weiterführende Informationen
Nach der frühen Nutzenbewertung durch das IQWiG entscheidet der G-BA über den Zusatznutzen. Sollte sich die AkdÄ mit einer Stellungnahme äußern, wird diese auf der AkdÄ-Website veröffentlicht.
Quelle
Europäischer Öffentlicher Beurteilungsbericht (EPAR) Veklury®, erschienen am 22. Dezember 2020. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Literatur
- Arzneimittelkommission der deutschen Ärzteschaft: Remdesivir (Veklury®). Arzneiverordnung in der Praxis (AVP) 2020; 47: 136-140.
- Kluge S, Janssens U, Welte T et al.: Empfehlungen zur stationären Therapie von Patienten mit COVID-19: www.awmf.org/uploads/tx_szleitlinien/113-001LGl_S3_Empfehlungen-zur-stationaeren-Therapie-von-Patienten-mit-COVID-19__2021-05.pdf (letzter Zugriff: 1. Juni 2021). AWMF-Registernummer: 113/001; Stand: 17. Mai 2021.
- Beigel JH, Tomashek KM, Dodd LE et al.: Remdesivir for the treatment of Covid-19 - final report. N Engl J Med 2020; 383: 1813-1826.
- WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection: A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis 2020; 20: e192-e197.
- WHO Solidarity Trial Consortium, Pan H, Peto R et al.: Repurposed antiviral drugs for Covid-19 - interim WHO Solidarity Trial results. N Engl J Med 2021; 384: 497-511.
- Wang Y, Zhang D, Du G et al.: Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020; 395: 1569-1578.
- Spinner CD, Gottlieb RL, Criner GJ et al.: Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA 2020; 324: 1048-1057.
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 8. Juni 2021 vorab online veröffentlicht.