Clopidogrel (Iscover®, Plavix®)
Neue Indikation: transitorische ischämische Attacke (TIA) und ischämischer Schlaganfall (IS)
Zugelassene Indikation und Wirkmechanismus
Iscover® und Plavix® (Clopidogrel) haben, in Kombination mit Acetylsalicylsäure (ASS), eine weitere Zulassung erhalten bei erwachsenen Patienten mit transitorischer ischämischer Attacke (TIA) mit mäßigem bis hohem Risiko (ABCD2-Score ≥ 4) oder mit leichtem ischämischem Schlaganfall (IS) (NIHSS2 ≤ 3) innerhalb von 24 Stunden nach TIA bzw. IS (Erläuterung der Scores siehe unten).
Clopidogrel ist ein Prodrug, das durch CYP450-Enzyme in den aktiven Metaboliten überführt wird. Der aktive Metabolit von Clopidogrel hemmt selektiv die Bindung von Adenosindiphosphat (ADP) an dem Thrombozytenrezeptor P2Y12. Hierdurch wird die ADP-abhängige Thrombozytenaggregation über den Glykoprotein-IIb/IIIa-Rezeptorkomplex verhindert. Die Blockade des P2Y12-Rezeptors ist irreversibel. Daher wird die Thrombozytenaggregation über fünf bis sieben Tage, bis zur Bildung neuer Thrombozyten, gehemmt.
Markteinführung
Iscover® und Plavix® (Clopidogrel) sind in verschiedenen Indikationen seit 1998 auf dem deutschen Markt und in dieser Indikation seit 29. Januar 2021 zugelassen.
Bewertung
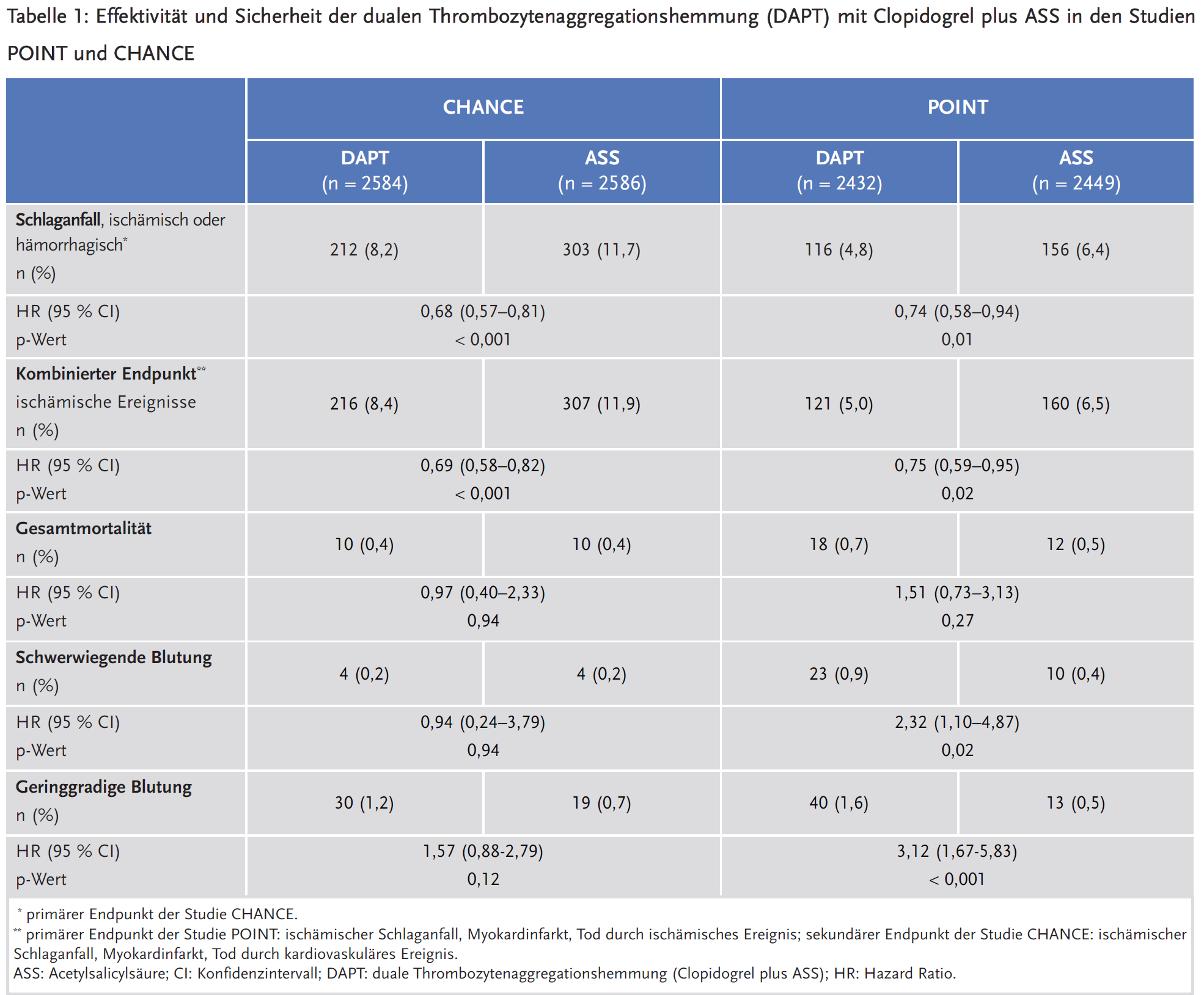
Die Zulassungsstudien CHANCE und POINT untersuchten die Effektivität und Sicherheit einer dualen Thrombozytenaggregationshemmung (DAPT) mit ASS plus Iscover® bzw. Plavix® (Clopidogrel) bei Patienten nach IS und TIA. Der Einschluss erfolgte innerhalb von 12 (POINT) bzw. 24 Stunden (CHANCE) nach Symptombeginn. Es wurden nur Patienten mit hohem Schlaganfallrisiko (IS oder TIA mit ABCD2-Score ≥ 4) und geringem Blutungsrisiko eingeschlossen, bei denen keine Indikation zur oralen Antikoagulation (OAK) bestand.
Die Vergleichsgruppe erhielt in beiden Studien eine Monotherapie mit ASS plus Placebo. In der Studie CHANCE erfolgte im Interventionsarm eine DAPT bis Tag 21 mit anschließender Clopidogrel-Monotherapie, in der Studie POINT wurde die DAPT dagegen bis Tag 90 fortgesetzt. Auch die Intensität der DAPT unterschied sich zwischen den Studien: In der Studie CHANCE erhielten alle Patienten eine fixe, niedrige ASS-Dosis von 75 mg, während in der Studie POINT knapp ein Viertel der Patienten ASS in einer Dosierung von mehr als 100 mg erhielt. Zudem war die Aufsättigungsdosis von Clopidogrel in der Studie POINT höher als in der Studie CHANCE (600 mg vs. 300 mg).
In beiden Studien wurde die Häufigkeit von Schlaganfällen (ischämisch oder hämorrhagisch) unter DAPT signifikant gesenkt. Die absolute Risikoreduktion bis Tag 90 betrug 1,6 % (POINT) bzw. 3,5 % (CHANCE), entsprechend einer Number needed to treat (NNT) von 63 bzw. 29. Blutungen waren überwiegend extrakraniell lokalisiert. Schwerwiegende Blutungen traten in der Studie POINT signifikant häufiger unter DAPT auf als im Kontrollarm (0,4 % vs. 0,9 %), in der Studie CHANCE bestand dagegen kein Unterschied zwischen den Studienarmen. Hämorrhagische Schlaganfälle waren in beiden Studien selten (CHANCE: n = 16, POINT: n = 8) und nicht signifikant gehäuft unter DAPT.
In der Ereigniszeitanalyse nahm der Effekt der DAPT auf ischämische vaskuläre Ereignisse mit zunehmender Dauer deutlich ab, während das Blutungsrisiko weitgehend konstant blieb. Die Europäische Arzneimittel-Agentur (EMA) schätzt das Nutzen-Risiko-Verhältnis der DAPT für eine Dauer von bis zu maximal 21 Tagen nach Symptombeginn als positiv ein. Allerdings zeigt sich in einer stratifizierten Analyse der Studie POINT, dass bereits ab der zweiten Behandlungswoche unter DAPT numerisch mehr schwere ischämische Ereignisse auftraten. Auch in der gepoolten Analyse der Studien erscheint ein positives Nutzen-Risiko-Verhältnis (Verhinderung schwerer ischämischer Ereignisse vs. zusätzliche schwere Blutungen) ab der zweiten Behandlungswoche unsicher. Aus Sicht der AkdÄ besteht deshalb nur bei niedrigem Blutungsrisiko und hohem Risiko für einen ischämischen Schlaganfall die Indikation für eine Verlängerung der DAPT über die erste Behandlungswoche hinaus. Entsprechend dem Protokoll der Studie CHANCE wird eine Aufsättigung mit 300 mg Clopidogrel und die Kombination mit niedrig dosiertem ASS (≤ 100 mg) empfohlen.
Wirksamkeit in den Zulassungsstudien
Nach IS oder TIA wird für Patienten ohne Vorhofflimmern zur Rezidivprophylaxe eine Thrombozytenaggregationshemmung empfohlen. Für die meisten Patienten ist die Therapie mit ASS Mittel der Wahl. Zwei randomisierte, doppelblinde Studien untersuchten, ob bei selektierten Patienten die zusätzliche Gabe von Clopidogrel gegenüber einer Monotherapie mit ASS vorteilhaft ist. Beide Studien wurden multizentrisch durchgeführt. Die Studie CHANCE (1) (n = 5170) fand ausschließlich in China statt, die Studie POINT (2) (n = 4881) überwiegend in den USA. Weniger als 10 % der Patienten kamen in der Studie POINT aus Europa.
Das Risiko, nach TIA einen Schlaganfall zu erleiden, kann mit Hilfe des ABCD2-Scores (0–7 Punkte) abgeschätzt werden. Der Score berücksichtigt bei der Risikokalkulation das Lebensalter, die Höhe des Blutdrucks, die Dauer und Art der Symptomatik sowie das Vorhandensein eines Diabetes mellitus. Bei einem ABCD2-Score von 4–5 Punkten hatten Patienten in Kohortenstudien ein Risiko von 10 %, in den folgenden 90 Tagen einen Schlaganfall zu erleiden (3). Die Schwere eines erlittenen Schlaganfalls wird mittels der NIH (National Institutes of Health) Stroke Skala (NIHSS) in einem Bereich von 0 bis 42 erfasst, wobei höhere Werte einer schwereren Symptomatik entsprechen. Es besteht eine starke Korrelation zwischen initialem NIHSS und per MRT ermitteltem Läsionsvolumen (4). Bei niedrigem NIHSS ist somit von einem kleinen ischämischem Infarktgebiet auszugehen.
In die Zulassungsstudien POINT und CHANCE wurden Patienten mit gering ausgeprägter Symptomatik nach IS (NIHSS ≤ 3) und Patienten mit TIA und moderat bis stark erhöhtem Risiko (ABCD2-Score ≥ 4) eingeschlossen, die weder durch ihre Begleitmedikation noch durch ihre Vorerkrankungen ein erhöhtes Blutungsrisiko hatten. Ausschlusskriterien waren deshalb unter anderem eine Indikation zur OAK (beispielsweise aufgrund von Vorhofflimmern) oder eine geplante Therapie mit NSAR über mehr als eine Woche, gastrointestinale Blutungen innerhalb von drei Monaten vor Studienteilnahme, jegliche nichttraumatische intrakranielle Blutung und bekannte Blutgerinnungsstörungen. Außerdem wurden Patienten mit einer Stenose der Arteria carotis interna (> 50 %) ausgeschlossen, wenn diese Stenose als mögliche Ursache der Symptomatik in Betracht kam. Der Studieneinschluss musste innerhalb von 12 Stunden (POINT) bzw. 24 Stunden (CHANCE) nach Symptombeginn erfolgen. Bei Patienten mit TIA durfte die Symptomatik nicht beschränkt sein auf isolierte Taubheitsgefühle, Sehstörungen oder Schwindel.
In beiden Studien erfolgte eine Randomisierung 1:1 zu Clopidogrel oder Placebo, jeweils zusätzlich zu ASS. In der Studie POINT wurde im Interventionsarm die duale Thrombozytenaggregationshemmung (DAPT) bis Tag 90 fortgeführt, in der Studie CHANCE dagegen nur bis Tag 21. Im Anschluss an die DAPT erhielten Patienten im Interventionsarm der Studie CHANCE eine Monotherapie mit Clopidogrel (plus Placebo-ASS) bis Tag 90. Zudem war die DAPT in der Studie CHANCE niedriger dosiert: Die Aufsättigungsdosis von Clopidogrel betrug 300 mg (vs. 600 mg in der Studie POINT) und ASS wurde nach einer initialen Aufsättigung (75–300 mg) in einer fixen Dosis von 75 mg gegeben, während in der Studie POINT ASS (ohne initiale Aufsättigung) in Dosierungen zwischen 50–325 mg gegeben wurde, bei knapp einem Viertel der Patienten in einer höheren Dosierung als 100 mg.
Die Patientencharakteristika waren zwischen den Armen ausgeglichen. Das Alter betrug durchschnittlich 62 Jahre (CHANCE) bzw. 65 Jahre (POINT). In der Studie POINT waren etwas mehr Frauen eingeschlossen (45 %) als in der Studie CHANCE (34 %). In beiden Studien war das Indexereignis häufiger ein IS, wobei dieser in der Studie CHANCE deutlicher überwog als in der Studie POINT (72 % vs. 57 %). Die durchschnittliche Symptomdauer vor Randomisierung betrug in der Studie POINT sieben Stunden, in der Studie CHANCE 13 Stunden.
Primärer Endpunkt der Studie CHANCE war die Häufigkeit von Schlaganfällen (ischämisch oder hämorrhagisch) an Tag 90. In der Studie POINT wurde primär ein kombinierter Endpunkt schwerer ischämischer Ereignisse (IS, Myokardinfarkt und Tod durch ein ischämisches vaskuläres Ereignis) bis Tag 90 untersucht, wobei die Einzelkomponenten sekundäre Endpunkte darstellten. Die Studie POINT wurde aufgrund einer Entscheidung des Datenüberwachungskomitees nach Einschluss von 84 % der ursprünglich geplanten Patienten gestoppt, weil ein signifikanter Behandlungsunterschied sowohl bezüglich des primären Wirksamkeitsendpunktes als auch des Sicherheitsendpunktes (schwerwiegende Blutung) erreicht worden war.
In beiden Studien wurde die Häufigkeit an Schlaganfällen (ischämisch oder hämorrhagisch) signifikant reduziert. In der Studie CHANCE zeigte sich dabei eine stärkere Risikoreduktion als in der Studie POINT. Das Risiko, innerhalb von 90 Tagen einen Schlaganfall zu erleiden, wurde in der Studie CHANCE durch DAPT um 3,5 % gesenkt (NNT 29), in der Studie POINT lediglich um 1,6 % (NNT 63). Der Unterschied zwischen den Studien war nicht statistisch signifikant (p-Wert für Interaktion: 0,56). Auffallend ist die insgesamt niedrige Ereignisrate in der Studie POINT (siehe Tabelle 1), insbesondere bei Patienten mit TIA (POINT: 4,4 % vs. CHANCE: 9,2 %). Die Autoren der Studie POINT vermuten als mögliche Ursache der niedrigen Ereignisrate den irrtümlichen Einschluss von Patienten mit nicht-ischämisch bedingter neurologischer Symptomatik, wie beispielsweise Migräne, epileptischen Anfällen oder psychiatrischen Erkrankungen. Die Behandlungsdifferenz des kombinierten Endpunktes beruhte in beiden Studien auf der Reduktion ischämischer Schlaganfälle; die übrigen Komponenten (Myokardinfarkt, Tod durch ischämisches bzw. kardiovaskuläres Ereignis) unterschieden sich nicht oder allenfalls numerisch minimal zwischen den Studienarmen.
Der Sicherheitsendpunkt „schwerwiegende Blutungen“ ist in beiden Studien ähnlich definiert. In der Studie CHANCE fallen darunter alle Blutungen mit Todesfolge, intrakranielle Blutungen sowie hämodynamisch wirksame Blutungen, die Transfusionen, inotrope Unterstützung oder einen chirurgischen Eingriff erfordern; in der Studie POINT werden nur symptomatische intrakranielle Blutungen als „schwerwiegend“ betrachtet, aber zusätzlich intraokulare Blutungen hinzugezählt, wenn diese zu einer Sehminderung führten. Während in der Studie CHANCE schwerwiegende Blutungen numerisch gleich häufig im Interventions- und Kontrollarm auftraten, war in der Studie POINT das Risiko für schwerwiegende Blutungen unter DAPT signifikant um mehr als das Doppelte erhöht. Die Mehrzahl der schwerwiegenden Blutungen trat extrakraniell auf. Hämorrhagische Schlaganfälle waren in beiden Studien selten (CHANCE: n = 16, POINT: n = 8) und nicht signifikant gehäuft unter DAPT. In der Studie POINT zeigte sich eine geringfügige, numerische Erhöhung der Gesamtmortalität unter DAPT; dieses Sicherheitssignal findet sich nicht in der Studie CHANCE (siehe Tabelle 1).
Die geringere Rate an Blutungen in der Studie CHANCE lässt sich am ehesten durch die kürzere Dauer der DAPT sowie die niedrigere Dosierung von ASS erklären. Zudem könnte eine Assoziation zwischen der niedrigen Blutungsrate und der erhöhten Prävalenz von Mutationen mit herabgesetzter Aktivität des CYP2C19-Enzyms bei chinesischen Patienten bestehen, da diese Clopidogrel unvollständig in seinen aktiven Metaboliten umwandeln. Eine Substudie von CHANCE fand allerdings kein signifikant unterschiedliches Blutungsrisiko zwischen Patienten mit und ohne CYP2C19-Varianten (5).
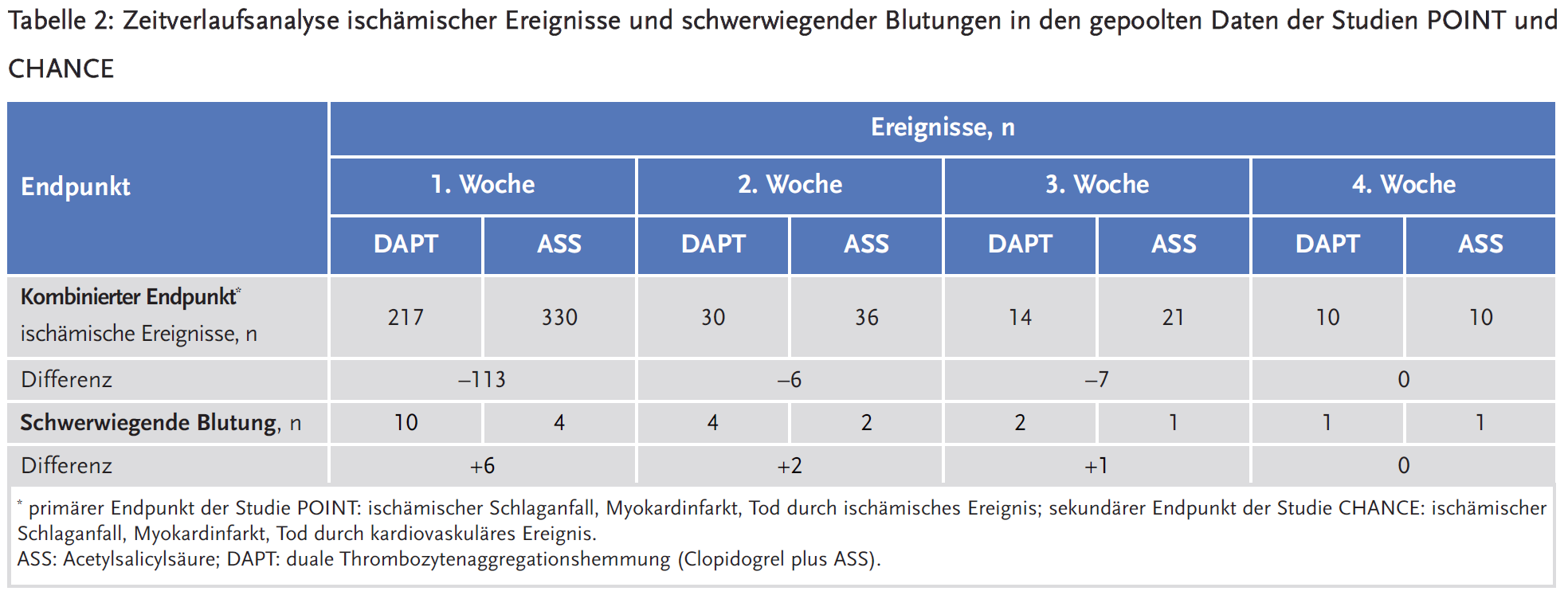
Der verlängerte Einsatz der DAPT in der Studie POINT reduzierte das Risiko ischämischer Ereignisse nicht stärker als die DAPT mit einer Dauer von 21 Tagen, wie sie in der Studie CHANCE zur Anwendung kam. In der Ereigniszeitanalyse nahm der Effekt der DAPT auf ischämische vaskuläre Ereignisse mit zunehmender Dauer deutlich ab, während das Blutungsrisiko über die Behandlungszeit hinweg weitgehend konstant blieb. Für die Studie POINT lag die mit Hilfe eines modellbasierten Ansatzes ermittelte optimale Behandlungsdauer bei 21 Tagen. Das Risiko, ein schweres ischämisches Ereignis (IS, Myokardinfarkt oder Tod durch ein ischämisches vaskuläres Ereignis) zu erleiden, wurde innerhalb der ersten 21 Tage durch DAPT um 35 % (Hazard Ratio [HR] 0,65; 95 % Konfidenzintervall [CI] 0,50–0,85; p = 0,0015) reduziert. In dem Zeitraum von 22 bis 90 Tagen nach Studieneinschluss war dieses Risiko dagegen um 38 % unter DAPT erhöht (HR 1,38; 95 % CI 0,81–2,35; p = 0,24). Basierend auf dieser Analyse wird das Nutzen-Risiko-Verhältnis der DAPT von der EMA für eine Dauer von bis zu maximal 21 Tagen nach Symptombeginn als positiv eingeschätzt.
Nach Einschätzung der AkdÄ ist dagegen ein positives Nutzen-Risiko-Verhältnis nur in der ersten Behandlungswoche belegt: Werden die Daten der Studien POINT und CHANCE gepoolt, so stehen in der ersten Woche 113 verhinderten schweren ischämischen Ereignissen sechs zusätzlich aufgetretene, schwerwiegende Blutungen gegenüber. Bereits in der zweiten Behandlungswoche verändert sich das Nutzen-Risiko-Verhältnis deutlich; hier stehen sechs verhinderten schweren ischämischen Ereignissen zwei zusätzliche schwerwiegende Blutungen gegenüber (siehe Tabelle 2). Hierzu konsistent zeigt sich in der Studie POINT ein gegensätzlicher Effektschätzer, wenn eine Analyse stratifiziert nach Tag 0–7 vs. Tag 8–90 erfolgt: Während in der ersten Behandlungswoche der primäre Endpunkt signifikant seltener unter DAPT erreicht wird (HR 0,74; 95 % CI 0,55–0,99), so erleiden im nachfolgenden Zeitraum numerisch mehr Patienten unter DAPT ein schwerwiegendes ischämisches Ereignis (HR 1,03, 95 % CI 0,70–1,53).
In der Studie CHANCE wurde im Interventionsarm nach Beendigung der DAPT die Thrombozytenaggregationshemmung mit Clopidogrel bis Tag 90 fortgeführt. In Deutschland ist dagegen die Monotherapie mit ASS Mittel der Wahl zur Sekundärprophylaxe nach IS oder TIA. Eine kurzzeitige DAPT mit anschließender ASS-Monotherapie beruht deshalb auf einer Extrapolation der Studienergebnisse von CHANCE.
Ausgewählte Nebenwirkungen
Die häufigsten Nebenwirkungen von Clopidogrel sind Blutungen. Pathophysiologische Erwägungen lassen vermuten, dass nach IS unter DAPT ein erhöhtes Risiko für eine hämorrhagische Transformation des Infarktgebietes besteht. In den Zulassungsstudien gibt es – bei hoch selektierten Patienten – keinen Beleg für diese Hypothese. In der Studie CHANCE traten intrazerebrale Blutungen gleich häufig in beiden Studienarmen auf, in der Studie POINT bestand unter DAPT ein numerisch geringfügig erhöhtes Risiko, das bei sehr kleinen Fallzahlen (n = 8) zufallsbedingt sein kann. Für weniger selektierte Patienten mit IS kann das Risiko intrazerebraler Blutungen unter DAPT aktuell nicht sicher beurteilt werden.
Ausgewählte Warnhinweise/Kontraindikationen/Interaktionen
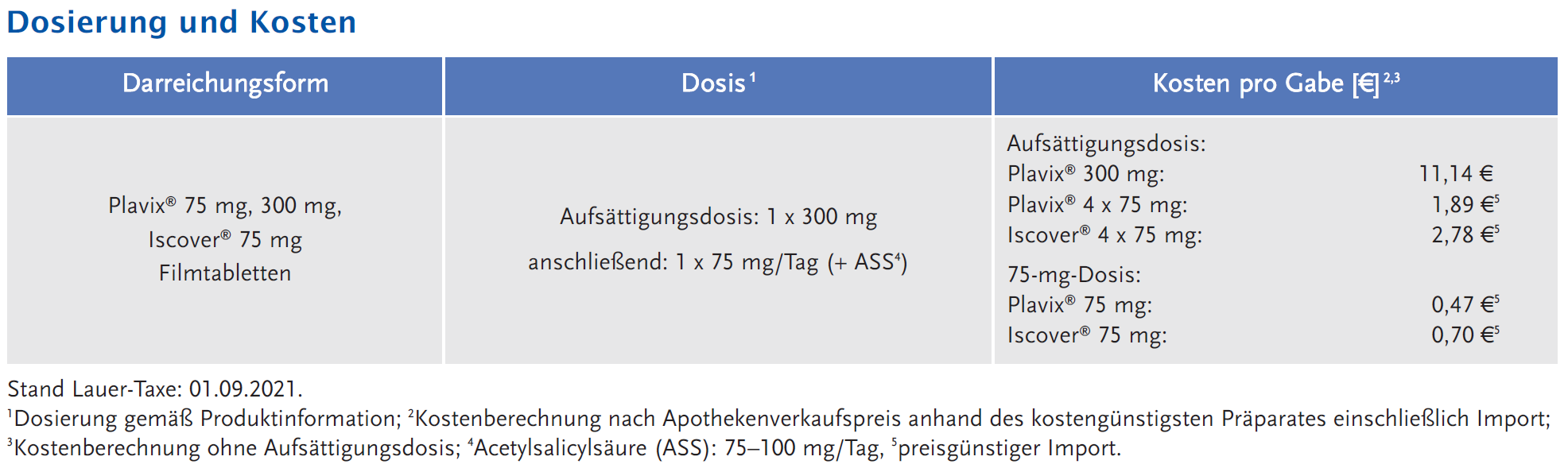
- Es gibt keine Hinweise, dass eine höhere Aufsättigungsdosis von Clopidogrel oder eine höhere Dosierung von ASS mit einer stärkeren Reduktion von IS assoziiert ist. Um Blutungsrisiken zu minimieren, wird deshalb entsprechend dem Protokoll der Studie CHANCE eine Aufsättigung mit 300 mg Clopidogrel und die Kombination mit niedrig dosiertem ASS (≤ 100 mg) empfohlen.
- Patienten ≥ 65 Jahren hatten in den gepoolten Daten ein doppelt so hohes Risiko für schwerwiegende Blutungen wie jüngere Patienten des gleichen Studienarms (ASS: 0,2 % vs. 0,5 %, DAPT: 0,4 % vs. 0,9 %). Die Risikosteigerung unter DAPT war dabei bei Patienten ≥ 65 Jahren geringfügig starker ausgeprägt als bei jüngeren Patienten (HR 1,67 vs. 1,50). Patienten ≥ 75 Jahre machten in den gepoolten Daten 19 % der eingeschlossenen Patienten aus. Für diese Patienten liegen jedoch keine Subgruppenanalysen vor. Bei Patienten ≥ 75 Jahren sollte besonders kritisch Nutzen und Risiko einer DAPT abgewogen werden und ihre Dauer möglichst kurz gewählt werden.
- Patienten mit schwerer Niereninsuffizienz waren aus den Studien POINT und CHANCE ausgeschlossen (POINT: Serumkreatinin > 2 mg/dL, CHANCE: nicht definiert). Es liegen insgesamt nur sehr wenige Daten zur Effektivität und Sicherheit von Clopidogrel bei Patienten mit Niereninsuffizienz vor. Eine kleine Studie zur Pharmakokinetik (n = 16) fand bei Patienten mit mittelgradig (Kreatinin-Clearance 30–60 ml/min) und stark eingeschränkter Nierenfunktion (5–15 ml/min) eine Verlängerung der Blutungszeit, die der Wirkung von Clopidogrel bei nierengesunden Patienten entsprach (6). Aufgrund der begrenzten Erfahrung wird empfohlen, Clopidogrel bei Patienten mit schwerer Niereninsuffizienz mit besonderer Vorsicht anzuwenden.
Weiterführende Informationen
Clopidogrel (Iscover®, Plavix®) wurde für diese Indikation nicht in die Bewertung des Zusatznutzens nach § 35a SGB V vom G-BA aufgenommen, da es bereits vor in Kraft treten des AMNOG 2011 auf dem deutschen Markt eingeführt wurde.
Quelle
Europäischer Öffentlicher Beurteilungsberichte (EPAR): Assessment Report – Variation Iscover®, Plavix®, erschienen am 12. Februar 2021. Die vorliegende Information erhebt keinen Anspruch auf Vollständigkeit. Für die Richtigkeit der angegebenen Dosierungen kann keine Gewähr übernommen werden.
Literatur
- Wang Y, Wang Y, Zhao X et al.: Clopidogrel with aspirin in acute minor stroke or transient ischemic attack. N Engl J Med 2013; 369: 11-19.
- Johnston SC, Easton JD, Farrant M et al.: Clopidogrel and Aspirin in Acute Ischemic Stroke and High-Risk TIA. N Engl J Med 2018; 379: 215-225.
- Johnston SC, Rothwell PM, Nguyen-Huynh MN et al.: Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. Lancet 2007; 369: 283-292.
- Tong DC, Yenari MA, Albers GW et al.: Correlation of perfusion- and diffusion-weighted MRI with NIHSS score in acute (<6.5 hour) ischemic stroke. Neurology 1998; 50: 864-870.
- Wang Y, Zhao X, Lin J et al.: Association between cyp2c19 loss-of-function allele status and efficacy of clopidogrel for risk reduction among patients with minor stroke or transient ischemic attack. JAMA 2016; 316: 70-78.
- Deray G, Bagnis C, Brouard R et al.: Clopidogrel activities in patients with renal function impairment. Clin Drug Investig 1998; 16: 319-328.
Hinweise
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 8. September 2021 vorab online veröffentlicht.