Arzneimittel: Sinnvolle Studien nach der Zulassung
Nachdruck aus: Deutsches Ärzteblatt 2021; 118: A 1148-1152
Die Arzneimittelkommission der deutschen Ärzteschaft erklärt, an welchen Postzulassungsstudien Ärztinnen und Ärzte teilnehmen sollten – und an welchen nicht.
Häufig sind nach der Zulassung eines Arzneimittels noch viele Fragen offen, die für die Gesamtbewertung des neuen Medikaments von großer Bedeutung sind. Denn die klinischen Studien, auf denen die Zulassung beruht, werden in der Regel über einen relativ kurzen Zeitraum mit einer begrenzten Anzahl von nach strengen Ein- und Ausschlusskriterien ausgewählten Patienten durchgeführt, die meist einem „durchschnittlichen“ Patienten in der Praxis nicht entsprechen (1). Anhand dieser Studien überprüfen die Arzneimittelbehörden im Zulassungsverfahren primär die Wirksamkeit und Unbedenklichkeit eines Arzneimittels (2). Als Indikator für die Wirksamkeit sind dabei häufig nicht validierte Surrogatendpunkte ausreichend für die Zulassung (3).
Daher fehlen nach der Zulassung oft Daten zu patientenrelevanten Endpunkten wie zur Mortalität oder gesundheitsbezogenen Lebensqualität, ebenso wie „Head-to-head“-Vergleiche mit anderen Therapieoptionen. Außerdem fehlen Daten zum Nutzen und Schaden eines Arzneimittels bei längerfristiger Anwendung und bei besonderen Patientengruppen wie beispielsweise älteren Patienten. Auch können seltene Nebenwirkungen, Wechselwirkungen oder andere Risiken im Zusammenhang mit der Arzneimittelanwendung in den klinischen Studien vor der Zulassung üblicherweise nicht erkannt werden (1). Die Ergebnisse der Zulassungsstudien sind zudem häufig nur eingeschränkt auf Patienten in Deutschland übertragbar, denn es handelt sich oft um international und multizentrisch durchgeführte Studien, die große regionale Unterschiede bei den Studienteilnehmern und bei den Studienergebnissen aufweisen (4). Darüber hinaus fehlt eine Bestätigung der Studienergebnisse, auf denen die Zulassung beruht. Besonders bei Arzneimitteln, die mit beschleunigter Zulassung auf den Markt gebracht wurden oder als Arzneimittel für seltene Leiden (Orphan Drug), besteht häufig nur eine schwache Evidenzbasis.
Sinnvolle Studienarten
Anwendungsbeobachtungen (AWB) können die klinisch relevanten offenen Fragen nach der Zulassung eines Arzneimittels nicht beantworten und werden häufig aus Marketinginteressen durchgeführt. Deshalb hat die Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) Kolleginnen und Kollegen von einer Teilnahme abgeraten (5). Es gibt jedoch Studien nach der Zulassung, die die Arzneimittelkommission ausdrücklich unterstützt. Dazu gehören von den Arzneimittelbehörden angeordnete „Post-Authorisation Safety Studies“ (PASS) und „Post-Authorisation Efficacy Studies“ (PAES) sowie die vom Gemeinsamen Bundesausschuss (G-BA) angeordneten anwendungsbegleitenden Datenerhebungen.
Nach der Zulassung kann Evidenz zu den offenen Fragen mit verschiedenen Studienarten und aus unterschiedlichen Datenquellen erhoben werden (siehe Tabelle 1). Die Wahl des geeigneten Studiendesigns richtet sich nach der Fragestellung, aber auch beispielsweise nach der Anzahl der betroffenen Patienten. Besonders relevant für die Beantwortung der offenen Fragen nach der Zulassung sind PASS und PAES, die von den regulatorischen Behörden, vor allem der Europäischen Arzneimittel-Agentur (EMA), im Rahmen beschleunigter Zulassungsverfahren zunehmend häufig angeordnet oder freiwillig von den pharmazeutischen Unternehmern (pU) durchgeführt werden, ebenso wie die neu eingeführten anwendungsbegleitenden Datenerhebungen (6-11).
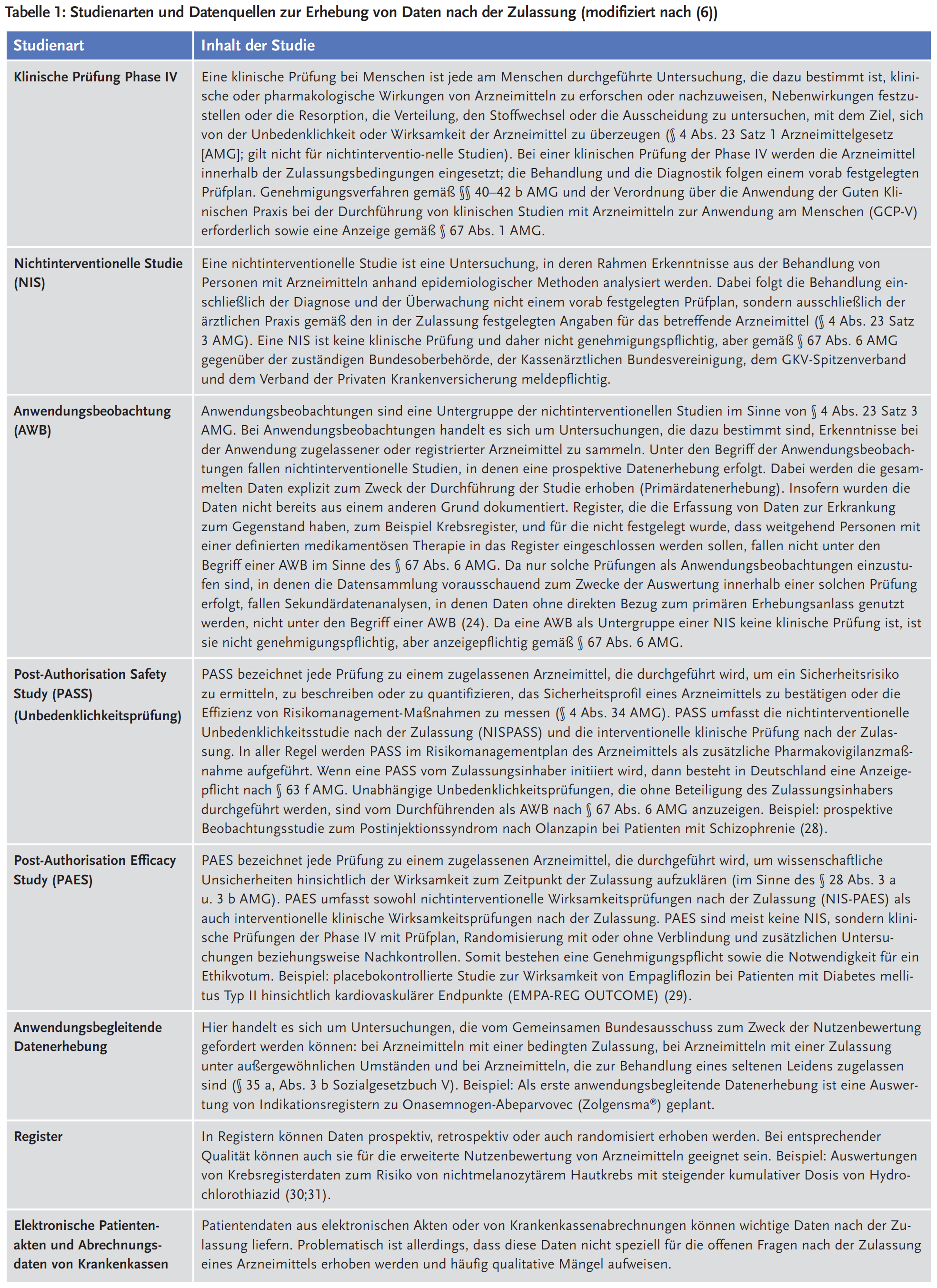
In einer PASS sollen Sicherheitsrisiken identifiziert, charakterisiert oder quantifiziert, das Sicherheitsprofil eines Arzneimittels bestätigt oder die Wirksamkeit von präventiven Maßnahmen des Risikomanagements überprüft werden (12). Eine PASS kann beispielsweise als Bedingung für die Zulassung unter außergewöhnlichen Umständen angeordnet werden oder wenn nach der Zulassung Sicherheitsbedenken auftreten. Das Design ist abhängig von der Fragestellung. PASS können sowohl als interventionelle, klinische Studie als auch als nichtinterventionelle Studie (NIS) durchgeführt werden. Möglich sind Kohorten- und Fall-Kontroll-Studien sowie Studien zur Arzneimittelanwendung („Drug Utilization Studies“), aber auch systematische Übersichtsarbeiten und Metaanalysen, Auswertungen von Registern oder Erhebungen zum Follow-up von Studienpatienten.
Fragen zur Wirksamkeit
In einer PAES werden gezielt Fragen zur Wirksamkeit untersucht. Eine PAES kann beispielsweise angeordnet werden, wenn die Zulassung auf Surrogatendpunkten beruht, wenn Daten für bestimmten Patientengruppen oder in der Langzeitanwendung fehlen (13;14). Das Design einer PAES wird in Abhängigkeit von der Fragestellung gewählt (15). Bevorzugt sollten randomisierte klinische Studien (RCT) durchgeführt werden. Bei bestimmten Arzneimitteln, zum Beispiel Impfstoffen, oder in bestimmten Situationen, beispielsweise wenn Endpunkte sehr selten auftreten, können auch nichtrandomisierte Studien mit paralleler oder historischer Kontrolle gerechtfertigt sein.
Im Gesetz für mehr Sicherheit in der Arzneimittelversorgung wurde festgelegt, dass der G-BA bei Arzneimitteln mit einer bedingten Zulassung („conditional marketing authorisation“), bei Arzneimitteln mit einer Zulassung unter außergewöhnlichen Umständen („approval under exceptional circumstances“) und bei Arzneimitteln zur Behandlung seltener Erkrankungen (Orphan Drugs) von den Herstellern anwendungsbegleitende Datenerhebungen fordern kann. Dabei kann es sich um unterschiedliche Studien handeln – RCT sind allerdings ausdrücklich ausgenommen (9). Arzneimittel für neuartige Therapien („Advanced Therapy Medicinal Products“, ATMP), wie zum Beispiel CAR-T-Zellen, werden im Gesetz nicht explizit erwähnt, sind jedoch meist Orphan Drugs, sodass für diese Arzneimittel deswegen anwendungsbegleitende Datenerhebungen angeordnet werden können.
Wenn der G-BA feststellt, dass eine anwendungsbegleitende Datenerhebung erforderlich ist, erstellt er oder das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) im Auftrag des G-BA einen Konzeptentwurf, in dem wesentliche Anforderungen zur Art, Dauer und Umfang der Datenerhebung, die Forschungsfrage, patientenrelevante Endpunkte sowie Informationen zur Methodik und Auswertung der Datenerhebung enthalten sind. Der Konzeptentwurf wird anschließend von den zuständigen Bundesoberbehörden, dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und dem Paul-Ehrlich-Institut (PEI), den wissenschaftlich-medizinischen Fachgesellschaften, der AkdÄ und gegebenenfalls weiteren Fachleuten wie Registerbetreibern schriftlich bewertet. Auch der pU hat die Möglichkeit, eine schriftliche Erklärung zum Konzeptentwurf abzugeben. Danach legt der G-BA die konkreten Vorgaben zur Durchführung der Datenerhebung fest, darunter auch die Zeitpunkte zur Überprüfung der Datenerhebung, die in regelmäßigen Abständen, mindestens jedoch alle 18 Monate, durch den G-BA erfolgt (10). Die anwendungsbegleitende Datenerhebung muss als klinische Prüfung dem Arzneimittelgesetz (AMG) genügen: Sie muss von einer Ethikkommission zustimmend bewertet und von der zuständigen Bundesoberbehörde genehmigt werden. Ärzte, die an den Studien nicht teilnehmen können oder wollen, kann der G-BA von der Verordnung dieser Arzneimittel ausschließen.
Indikationsregister verwendet
Das Gentherapeutikum Onasemnogen-Abeparvovec (Zolgensma®) ist das erste Arzneimittel, bei dem anwendungsbegleitende Daten erhoben und ausgewertet werden sollen. Es wurde von der EMA zur Behandlung von Säuglingen und Kleinkindern mit spinaler Muskelatrophie bedingt zugelassen. Im Unterschied zu Nusinersen (Spinraza®), dem bisher einzigen in dieser Indikation zugelassenen Arzneimittel, soll es in der Lage sein, die Erkrankung zu heilen – und das mit einer einzigen Anwendung.
Auf der Basis eines IQWiG-Konzepts, das auch von der AkdÄ kommentiert wurde, hat der G-BA die Anforderungen an die anwendungsbegleitende Datenerhebung definiert. Vorgesehen ist, dass mithilfe von Indikationsregistern möglichst 500 Kinder mit präsymptomatischer spinaler Muskelatrophie und mit spinaler Muskelatrophie Typ 1 und 2 in die Datenerhebung eingeschlossen werden. Die Therapie mit Onasemnogen-Abeparvovec wird dabei mit der Anwendung von Nusinersen verglichen. Ein besonderer Schwerpunkt der Datenauswertung soll auf Todesfälle, Krankheitsverläufe inklusive schwerer Komplikationen und Nebenwirkungen gelegt werden. Über den langfristigen Zusatznutzen von Onasemnogen-Abeparvovec wird der G-BA spätestens ab Sommer 2027 beraten.
Eine anwendungsbegleitende Datenerhebung ist nicht mit einer Anwendungsbeobachtung gleichzusetzen. Bei einer anwendungsbegleitenden Datenerhebung wird vergleichende Evidenz für die Nutzenbewertung erhoben. Ziel ist es, eine valide Quantifizierung des Zusatznutzens zu erreichen. Festgelegt werden auch gegebenenfalls notwendige anwendungsbegleitende Interventionen, wie beispielsweise bestimmte diagnostische Maßnahmen. Bei einer AWB sollen dagegen Daten nur durch Beobachtung der Anwendung erhoben werden. Laut AMG folgt die Behandlung einschließlich der Diagnose und Überwachung nicht einem Prüfplan, sondern ausschließlich der ärztlichen Praxis.
Anforderungen einhalten
Zur Beantwortung der offenen Fragen nach der Zulassung können abhängig von der Fragestellung verschiedene Studienarten und Datenquellen sinnvoll sein. Durchgängig müssen dabei allerdings grundlegende Anforderungen an klinische Studien eingehalten werden. Dazu gehören beispielsweise eine Formulierung der Fragestellung nach dem PICO-Schema (PICO: Akronym aus Patienten, Intervention, Comparator, Outcome) und ein vor Beginn der Datenerhebung finalisiertes Studienprotokoll mit einem präspezifizierten Analyseplan (11).
Die AkdÄ empfiehlt ausdrücklich die Teilnahme an PAES/PASS, die von den Arzneimittelbehörden angeordnet wurden. Aus Sicht der AkdÄ ist allerdings problematisch, dass pU angeordnete Studien häufig gar nicht durchführen (16-18): Etwa die Hälfte der angeordneten Studien wird nicht fristgerecht (19) und teils erst innerhalb von fünf bis sechs Jahren abgeschlossen (20). Die Ergebnisse der durchgeführten Studien bestätigen nur selten den anfangs propagierten Zusatznutzen der neuen Arzneimittel (21). Wenn pU den Auflagen zur Durchführung von Studien nicht nachkommen, wird das von Arzneimittelbehörden bisher nicht ausreichend sanktioniert. Die AkdÄ empfiehlt daher auch, pU gesetzlich zur Generierung von Daten nach der Zulassung zu verpflichten. Beispielsweise könnte die Anerkennung des Zusatznutzens durch den G-BA an die Erhebung weiterer Daten gebunden und befristet werden.
Dagegen rät die AkdÄ von der Teilnahme an AWB ab, die nicht von Arzneimittelbehörden oder G-BA angeordnet wurden, weil es sich oftmals um Marketingmaßnahmen ohne wissenschaftlichen Anspruch handelt (5;22). Verschiedene Untersuchungen stellen den rein beobachtenden Charakter von AWB infrage und deuten darauf hin, dass sie das Verordnungsverhalten von Ärztinnen und Ärzten verändern können (5;23). Obwohl AWB vielfach kritisiert wurden, findet sich der Begriff unglücklicherweise in der Verfahrensordnung des G-BA zur anwendungsbegleitenden Datenerhebung (9). Die AkdÄ empfiehlt, dass von Arzneimittelbehörden oder dem G-BA angeordnete Studien eindeutig gekennzeichnet werden, um die Entscheidung über eine Teilnahme zu erleichtern.
Evidenzlage verbessern
Die AkdÄ empfiehlt ebenso ausdrücklich die Teilnahme an anwendungsbegleitenden Datenerhebungen. Deren Einführung ist eine sinnvolle Entwicklung, die laut Gesetz jedoch auf bedingt zugelassene Arzneimittel, Arzneimittel mit einer Zulassung unter außergewöhnlichen Umständen und Orphan Drugs beschränkt ist. Wünschenswert wäre, diese Option auch für andere Arzneimittel zu öffnen. Eine anwendungsbegleitende Datenerhebung sollte nicht erst mit der Zulassung, sondern frühzeitig im Entwicklungsprozess eines Arzneimittels geplant werden, um beispielsweise Register anzupassen. Manche offenen Fragen nach der Zulassung können auch nur durch RCT beantwortet werden. Es ist daher unverständlich, warum diese für die anwendungsbegleitende Datenerhebung ausdrücklich ausgenommen werden (25). Die AkdÄ hat bereits 2018 in einer gemeinsamen Stellungnahme mit der Bundesärztekammer festgestellt, dass vor allem gut konzipierte und durchgeführte RCT oder zumindest prospektive vergleichende Kohortenstudien benötigt werden, um die Evidenzlage für diese Arzneimittel zu verbessern, die nicht nur begonnen, sondern auch abgeschlossen und veröffentlicht werden (26).
Die AkdÄ fordert darüber hinaus mehr Studien, die unabhängig von pU durchgeführt werden. PU haben oft kein Interesse an den offenen Fragen nach der Zulassung eines Arzneimittels. Für unabhängige Forschung könnte ein Fonds eingerichtet werden, der beispielsweise durch Staat, Krankenversicherungen und pU finanziert wird. Alternativ oder zusätzlich könnte ein Modell wie in Italien eingeführt werden: Dort müssen pU fünf Prozent ihres Marketingbudgets in einen Fonds einzahlen, aus dem dann unabhängige, versorgungsnahe Forschung finanziert wird (27). Die vom Bundesforschungsministerium aufgelegten Förderprogramme für klinische Studien sind zwar zu begrüßen, aber von der budgetären Ausstattung her nicht geeignet, eine relevante Änderung zugunsten unabhängiger klinischer Studien zu erwirken.
Literatur
- Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM): Pharmakovigilanz: www.bfarm.de/DE/Arzneimittel/Pharmakovigilanz/_node.html. Letzter Zugriff: 26. Januar 2021.
- Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM): Arzneimittelzulassung: www.bfarm.de/DE/Buerger/Arzneimittel/Arzneimittelzulassung/_node.html. Letzter Zugriff: 26. Januar 2021.
- Surrogate patientenrelevanter Endpunkte. In: Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) (Hrsg.). Allgemeine Methoden: www.iqwig.de/methoden/allgemeine-methoden_version-6–0.pdf (letzter Zugriff: 11. März 2021). Version 6.0 vom 5. November 2020; 44-46.
- Können therapeutische Ergebnisse großer internationaler Arzneimittelstudien auf Patienten in Mitteleuropa übertragen werden? Arzneimittelbrief 2020; 54: 69.
- Schott G, Ludwig W-D, Lieb K: Anwendungsbeobachtungen: Erkenntnisgewinn ist gering. Dtsch Arztebl 2020; 117: A 1380-1381.
- Wem nutzen Anwendungsbeobachtungen? Arzneimittelbrief 2017; 51: 48DB01.
- Ludwig W-D: Klinische Endpunkte in Studien: Was ist relevant für HTA und Patienten? Forum 2020; 35: 368-372.
- Kaiser T, Wieseler B: Welche Anwendungsdaten brauchen wir für einen besseren Wissenstransfer? Forum 2020; 35: 284-288.
- Müller T, Bundesministerium für Gesundheit (BMG) an den Gemeinsamen Bundesausschuss (G-BA): Beschluss des Gemeinsamen Bundesausschusses gemäß § 91 SGB V vom 16. Juli 2020, hier: Änderung der Verfahrensordnung (VerfO): Änderung im 5. Kapitel –Verfahren zur Forderung einer anwendungsbegleitenden Datenerhebung nach § 35a Absatz 3b SGB V: www.g-ba.de/downloads/40-268-6988/2020-07-16_VerfO_Kapitel-5_BMG.pdf (letzter Zugriff: 26. Januar 2021). Berlin, 26. Oktober 2020.
- Gemeinsamen Bundesausschuss (G-BA): G-BA konkretisiert Verfahren zu anwendungsbegleitender Datenerhebung – Gentherapie Zolgensma erster Fall: www.g-ba.de/presse/pressemitteilungen/874/ (letzter Zugriff: 26. Januar 2021). Pressemitteilung vom 16. Juli 2020.
- Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG): [A19–43] Wissenschaftliche Ausarbeitung von Konzepten zur Generierung versorgungsnaher Daten und deren Auswertung zum Zwecke der Nutzenbewertung von Arzneimitteln nach § 35a SGB V– Rapid Report: www.iqwig.de/projekte/a19-43.html (letzter Zugriff: 26. Januar 2021). Stand: 14. Mai 2020.
- European Medicines Agency (EMA): Post-authorisation safety studies (PASS): www.ema.europa.eu/en/human-regulatory/post-authorisation/pharmacovigilance/post-authorisation-safety-studies-pass-0. Letzter Zugriff: 26. Januar 2021.
- European Medicines Agency (EMA): Post-authorisation efficacy studies: questions and answers: www.ema.europa.eu/en/human-regulatory/post-authorisation/post-authorisation-efficacy-studies-questions-answers. Letzter Zugriff: 26. Januar 2021.
- The European Commission (EC): Commission delegated regulation (EU) No 357/2014 of 3 February 2014 supplementing Directive 2001/83/EC of the European Parliament and of the Council and Regulation (EC) No 726/2004 of the European Parliament and of the Council as regards situations in which post-authorisation efficacy studies may be required. Official Journal of the European Union 2014: L107/101-L107/104.
- European Medicines Agency (EMA): Scientific guidance on post-authorisation efficacy studies: www.ema.europa.eu/en/documents/scientific-guideline/scientific-guidance-post-authorisation-efficacy-studies-first-version_en.pdf (letzter Zugriff: 11. März 2021). London, 12. Oktober 2016.
- Wieseler B, McGauran N, Kaiser T: New drugs: where did we go wrong and what can we do better? BMJ 2019; 366: l4340.
- Cipriani A, Ioannidis JPA, Rothwell PM et al.: Generating comparative evidence on new drugs and devices after approval. Lancet 2020; 395: 998-1010.
- Vreman RA, Leufkens HGM, Kesselheim AS: Getting the right evidence after drug approval. Front Pharmacol 2020; 11: 569535.
- Hoekman J, Klamer TT, Mantel-Teeuwisse AK et al.: Characteristics and follow-up of postmarketing studies of conditionally authorized medicines in the EU. Br J Clin Pharmacol 2016; 82: 213-226.
- Woloshin S, Schwartz LM, White B, Moore TJ: The fate of FDA postapproval studies. N Engl J Med 2017; 377: 1114-1117.
- Pease AM, Krumholz HM, Downing NS et al.: Postapproval studies of drugs initially approved by the FDA on the basis of limited evidence: systematic review. BMJ 2017; 357: j1680.
- Spelsberg A, Prugger C, Doshi P et al.: Contribution of industry funded post-marketing studies to drug safety: survey of notifications submitted to regulatory agencies. BMJ 2017; 356: j337.
- Koch C, Schleeff J, Techen F et al.: Impact of physicians‘ participation in non-interventional post-marketing studies on their prescription habits: A retrospective 2-armed cohort study in Germany. PLoS Med 2020; 17: e1003151.
- Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM): Gemeinsame Empfehlungen des Bundesinstituts für Arzneimittel und Medizinprodukte und des Paul-Ehrlich-Instituts zu Anwendungsbeobachtungen nach § 67 Absatz 6 Arzneimittelgesetz und zur Anzeige von nichtinterventionellen Unbedenklichkeitsprüfungen nach § 63f Arzneimittelgesetz: www.bfarm.de/SharedDocs/Downloads/DE/Arzneimittel/Zulassung/klin-pr/nichtInterventPruef/Gemeinsame%20Empfehlungen%20zu%20AWB%20und%20PASS.html (letzter Zugriff: 11. März 2021). Bonn, 20. Dezember 2019.
- Stegmaier P, Windeler J: „Ohne Vergleich geht gar nichts“. Monitor Versorgungsforschung 2019; Ausgabe 6: 32-35.
- Bundesärztekammer, Arzneimittelkommission der deutschen Ärzteschaft: Gemeinsame Stellungnahme der Bundesärztekammer und der Arzneimittelkommission der deutschen Ärzteschaft zum Referentenentwurf eines Gesetzes für mehr Sicherheit in der Arzneimittelversorgung (GSAV) (Bearbeitungsstand: 14.11.2018). Berlin, 14. Dezember 2018.
- Italian Medicines Agency (AIFA): Independent clinical research: www.aifa.gov.it/en/web/guest/ricerca-clinica-indipendente. Letzter Zugriff: 26. Januar 2021.
- Meyers KJ, Upadhyaya HP, Landry JL et al.: Postinjection delirium/sedation syndrome in patients with schizophrenia receiving olanzapine long-acting injection: results from a large observational study. BJPsych Open 2017; 3: 186-192.
- Zinman B, Wanner C, Lachin JM et al.: Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373: 2117-2128.
- Pottegard A, Hallas J, Olesen M et al.: Hydrochlorothiazide use is strongly associated with risk of lip cancer. J Intern Med 2017; 282: 322-331.
- Pedersen SA, Gaist D, Schmidt SAJ et al.: Hydrochlorothiazide use and risk of nonmelanoma skin cancer: A nationwide case-control study from Denmark. J Am Acad Dermatol 2018; 78: 673-681.
Fußnote
1weitere Mitglieder des Fachausschusses:
Prof. Dr. med. Christopher Baethge, Prof. Dr. Johannes Köbberling, Prof. Dr. Thomas Lempert,
Prof. Dr. med. Wolf-Dieter Ludwig, Prof. Dr. Bruno Müller-Oerlinghausen, Dr. med. Birke Schneider