Romosozumab (Evenity®) ▼
(frühe Nutzenbewertung)
In Kürze
- Romosozumab über zwölf Monate gefolgt von Alendronsäure ist zugelassen für die Behandlung der manifesten Osteoporose bei postmenopausalen Frauen mit deutlich erhöhtem Frakturrisiko.
- Während das IQWiG zunächst einen beträchtlichen Zusatznutzen sah (im Vergleich zu Alendronsäure alleine), bemängelte die AkdÄ die geringe absolute Risikoreduktion und Unsicherheiten bezüglich kardialer und vaskulärer Nebenwirkungen.
- Dieser Kritik schloss sich das IQWiG an aufgrund nachträglicher Auswertungen von atypischen Oberschenkelfrakturen und kardio- und zerebrovaskulären Ereignissen.
- Der G-BA beschloss einen Hinweis auf einen geringen Zusatznutzen.
Romosozumab hemmt den Osteozyten-Botenstoff Sklerostin, der negativ die Knochenbildung reguliert. Er bewirkt die Aktivierung von Saumzellen, die Produktion von Knochenmatrix durch Osteoblasten sowie die Rekrutierung von Osteoprogenitorzellen; insgesamt wird also der Knochenaufbau verstärkt. Zusätzlich hemmt Romosozumab den Knochenabbau durch Verringerung der Expression von Osteoklastenmediatoren. Der resultierende Anstieg trabekulärer und kortikaler Knochenmasse verbessert Struktur und Festigkeit der Knochen.
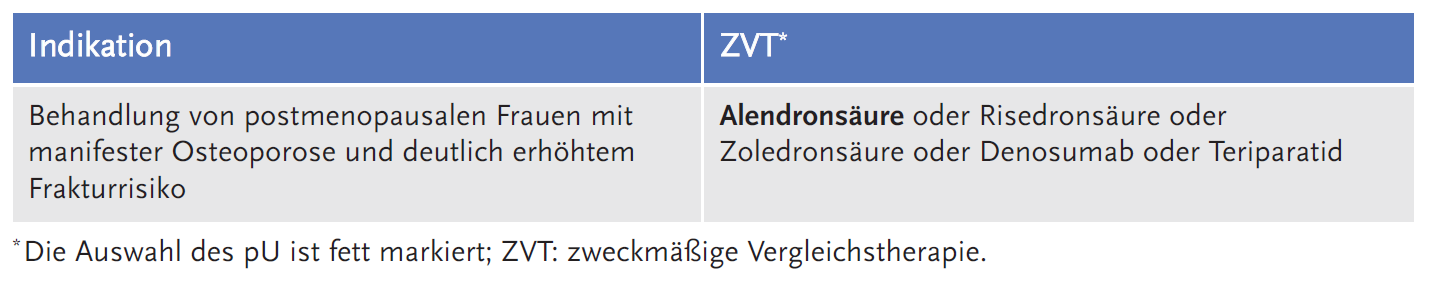
Der Gemeinsame Bundesausschuss (G-BA) legte die folgende Fragestellung fest und definierte dazu die zweckmäßige Vergleichstherapie (ZVT):
Vorgelegte Evidenz
Für die Nutzenbewertung legte der pU die Studie ARCH vor (1), eine multizentrische, doppelblinde, randomisierte Studie, in der die Wirkung von Romosozumab über zwölf Monate, gefolgt von zwölf Monaten Alendronsäure, mit Alendronsäure über 24 Monate alleine verglichen wurde. Eingeschlossen wurden 4093 postmenopausale Frauen, die mindestens eines der folgenden Kriterien bezüglich der Knochenmineraldichte (bone mineral density, BMD) und Frakturen erfüllten:
- BMD T-Score ≤ −2,5 an Hüfte oder Oberschenkelhals und entweder mindestens eine mittlere oder schwere vertebrale Fraktur oder mindestens zwei leichte vertebrale Frakturen oder
- BMD T-Score ≤ −2,0 an Hüfte oder Oberschenkelhals und entweder mindestens zwei mittlere oder schwere vertebrale Frakturen oder eine Fraktur des proximalen Oberschenkels, die innerhalb von 3 bis 24 Monaten vor der Randomisierung aufgetreten war.
Romosozumab senkte im Vergleich zur Alendronsäure statistisch signifikant das Risiko neuer vertebraler sowie klinisch relevanter Frakturen. Bezüglich Sicherheit, d. h. Nebenwirkungen (auch schwerwiegende), Therapieabbruch wegen Nebenwirkungen, Osteonekrose des Kiefers und Erkrankungen des Gastrointestinaltrakts – zeigten sich keine statistisch signifikanten Unterschiede zwischen den Behandlungsarmen. Verwertbare Daten zur Beeinflussung der gesundheitsbezogenen Lebensqualität wurden nicht vorgelegt.
Dossierbewertung des IQWiG
Das IQWiG kritisierte, dass die in der Studie ARCH definierten Kriterien für das Vorliegen eines deutlich erhöhten Frakturrisikos enger gefasst waren als in der Leitlinie des Dachverbandes Osteologie (DVO) (2), sodass die Studienpopulation von ARCH nicht alle Patientinnen abbildet, für die eine medikamentöse Therapie laut DVO-Leitlinie angezeigt ist. Allerdings wurde vom IQWiG die Diskrepanz zwischen dem Registereintrag zur Studie und den Daten im Modul 4A zum Endpunkt Osteonekrose des Kiefers aufgrund der geringen Ereignisrate nicht als relevant für die Nutzenbewertung eingeschätzt. In der Gesamtschau ergaben sich für das IQWiG vorwiegend positive Effekte für Romosozumab gegenüber Alendronsäure, zunächst resultierend in einem Hinweis auf einen beträchtlichen Zusatznutzen für Rosomozumab (3).
Stellungnahme der AkdÄ
Die AkdÄ stellte dagegen für Rosomozumab gegenüber Alendronsäure einen höchstens geringen Zusatznutzen fest. Dies beruhte auf der geringen absoluten Risikoreduktion für Hüft- und Beckenfrakturen (1 %) bzw. aller klinisch relevanten Frakturen (3 %) und auf dem Fehlen von Daten zur gesundheitsbezogenen Lebensqualität und zur kardialen und vaskulären Sicherheit (4).
Zudem wies die AkdÄ darauf hin, dass ein möglicher Zusatznutzen für Romosozumab allenfalls gegenüber Alendronsäure, nicht aber gegenüber Denosumab oder Teriparatid bestünde, da hierzu keinerlei belastbare Daten vorgelegt wurden. Auch wenn diese Antiosteoporotika ähnliche Frakturrisikoreduktion aufweisen, haben sie unterschiedliche Nebenwirkungen und damit einen unterschiedlichen Stellenwert in der Osteoporosebehandlung.
Nach der mündlichen Anhörung forderte der G-BA für die Bewertung des Zusatznutzens weitere Auswertungen. Das daraufhin angefertigte Addendum des IQWiG berücksichtigte u. a. die Endpunkte atypische Oberschenkelfraktur (ohne Unterteilung in symptomatisch/asymptomatisch), Nebenwirkungen, kardiale und zerebrovaskuläre Ereignisse sowie die Auswertung des Instruments Osteoporosis Assessment Questionnaire - Short Version (OPAQ-SV). Dabei ergab sich aufgrund der zerebrovaskulären Ereignisse ein Hinweis auf einen höheren Schaden durch Romosozumab. Zusammenfassend blieb lediglich ein Hinweis auf einen geringen Zusatznutzen gegenüber der ZVT bestehen (5).
Beschluss des G-BA
Abschließend stellte der G-BA die Verringerung relevanter Frakturen den zerebrovaskulären Nebenwirkungen in der Behandlung der manifesten Osteoporose bei postmenopausalen Frauen mit deutlich erhöhtem Frakturrisiko gegenüber (6). Beschlossen wurde ein Hinweis auf einen geringen Zusatznutzen von Romosozumab (gefolgt von Alendronsäure) gegenüber Alendronsäure alleine (Tabelle 1).

Literatur
- Saag KG, Petersen J, Brandi ML et al.: Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med 2017; 377: 1417-1427.
- Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e. V. (AWMF): Leitlinie des Dachverbands der Deutschsprachigen Wissenschaftlichen Osteologischen Gesellschaften e.V.: Prophylaxe, Diagnostik und Therapie der Osteoporose bei postmenopausalen Frauen und bei Männern: www.awmf.org/uploads/tx_szleitlinien/183-001l_S3_Osteoporose-Prophylaxe-Diagnostik-Therapie_2019-02.pdf (letzter Zugriff: 25. Juni 2020). AWMF-Register-Nummer: 183/001. Langversion 2017, Stand: 21. Februar 2019.
- IQWiG Dossierbewertung: www.g-ba.de/downloads/92-975-3591/2020-03-15_Nutzenbewertung-IQWiG_Romosozumab_D-516.pdf
- Stellungnahme der AkdÄ: www.akdae.de/Stellungnahmen/AMNOG/A-Z/Romosozumab/Romosozumab-EB.pdf
- IQWiG Addendum: www.g-ba.de/downloads/92-975-3775/2020-09-03_Addendum-IQWiG_Romosozumab_D-516.pdf
- G-BA: Tragende Gründe: www.g-ba.de/downloads/40-268-6825/2020-09-03_AM-RL-XII_Romosozumab_D-516_TrG.pdf
Hinweise
Arzneimittel, die mit einem schwarzen Dreieck (▼) gekennzeichnet sind, unterliegen einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelassenen Arzneimitteln oder zu neu zugelassenen Indikationen. Ziel ist es, den Ärzten zeitnah Informationen zu diesen Arzneimitteln zur Verfügung zu stellen, zunächst bei Markteinführung sowie nach der frühen Nutzenbewertung durch den Gemeinsamen Bundesausschuss (G-BA) (§ 35a Absatz 1 SGB V). „Neue Arzneimittel“ bei Markteinführung enthält Informationen basierend auf dem Europäischen Öffentlichen Bewertungsbericht (EPAR) der Europäischen Arzneimittel-Agentur (EMA) sowie weiteren bei Markteinführung vorliegenden Daten aus klinischen Studien. Nach Abschluss der frühen Nutzenbewertung wird der Zusatznutzen des neuen Arzneimittels und seine therapeutische Bedeutung auf der Basis der Dossierbewertung des IQWiG, der Stellungnahme der AkdÄ und des Beschlusses des G-BA im Rahmen der frühen Nutzenbewertung dargestellt („Update – Neue Arzneimittel“).
vorab online
Dieser Artikel wurde am 12. Oktober 2020 vorab online veröffentlicht.